|
Professor Department of Microbiology and Immunology Center for Infectious Diseases Ph.D., Temple University, 1984 Fellow American Academy of Microbiology Fellow American Association for the Advancement of Science |
||
|
|
E-mail: erich.mackow@stonybrook.edu |
||
|
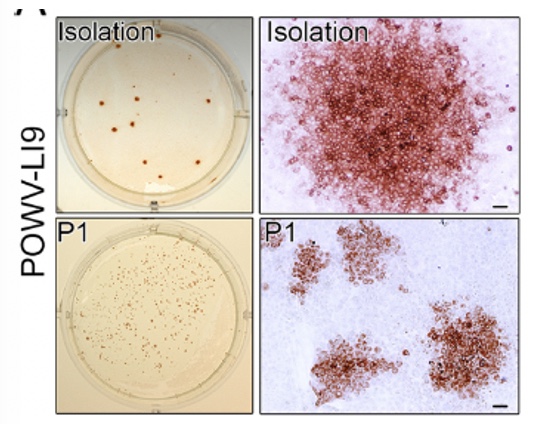
Research Studies of Molecular and Cellular Mechanisms of Viral Pathogenesis and Immune Evasion to Develop Novel Therapeutics and Vaccines Our lab investigates mechanisms of Flavivirus (FV: Powassan virus, Zika virus, Dengue virus), SARS-CoV2 and Hantavirus (HV) pathogenesis. One focus is on viral regulation of vascular endothelial cell (EC) functions that dynamically control pulmonary edema and permeability of the blood brain barrier (BBB). We further analyze mechanisms of POWV neuroinvasion via the blood CSF barrier (BCSFB) using in vitro and in vivo approaches. We study CNS cell responses that direct spongiform encephalitis, age-dependent lethality and long term neurologic damage. Roles for innate and adaptive responses in viral persistence and pathogenesis are being evaluated for use as attenuation and therapeutic targets. We developed a lethal age-dependent Powassan virus (POWV) murine model that permits analysis of POWV directed CNS damage, age-dependent neurovirulence mechanisms and potential therapeutic approaches to preventing POWV neurodegeneration and long-term neurologic sequelae. Our POWV reverse genetics system permits us to compare WT and attenuated POWVs, and define determinants of POWV spread, neuroinvasion and CNS pathology. These approaches reveal attenuating mutations for inclusion in POWV vaccines and mechanisms of POWV neurotropism, CNS damage and lethality. Within the ABSL3 we are using scRNAseq to analyze POWV-directed CNS responses that protect young mice, confer lethality to aged mice and define roles for senescent cells in CNS pathology. Powassan Virus Pathogenesis Isolation of Tick-Borne POWV L19: POWV-LI9 was isolated from Ixodes scapularis ticks (Deer ticks) by direct growth in VeroE6 cells. LI9 infected cells were uniquely found to spread focally cell-to-cell without cell lysis and in the presence of overlaid neutralizing antibodies. Cell-to-cell spread of LI9 is observed in epithelial cells, BHK21 and fibroblasts, but not during LI9 infection of ECs or pericytes, suggesting a cell type specific restriction. The mechanism of cell-to-cell spread immune evasion and cell specificity remain to be resolved and assayed for roles in pathogenesis.
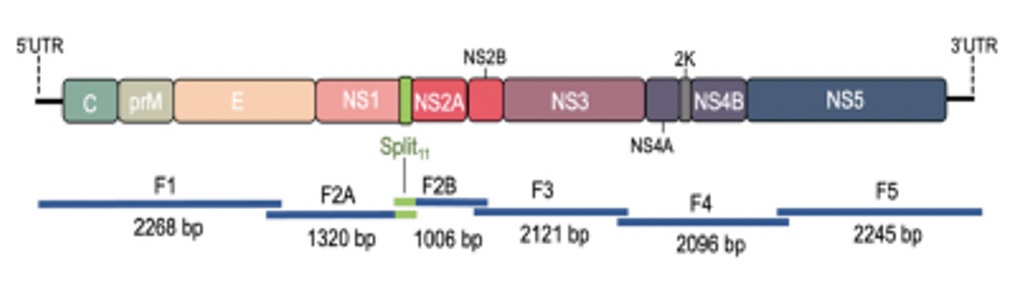
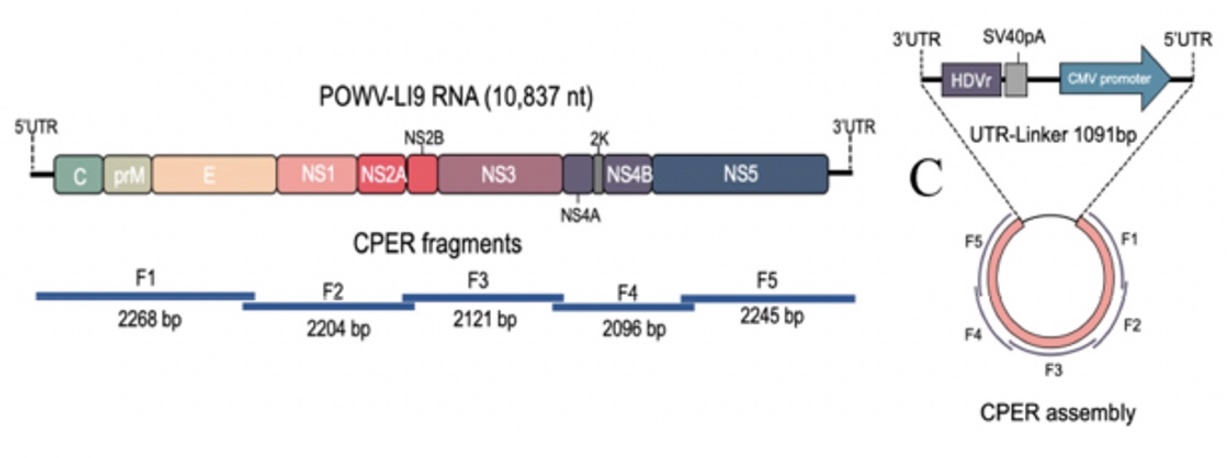 POWV Reverse Genetics: We developed a POWV reverse genetics system that uniquely permits us to modify infectious POWVs. Using a circular polymerase extension reaction (CPER), we generated recombinant LI9 (recLI9) POWVs, initially defining attenuating NS1 protein mutations and generating a recLI9-split-eGFP reporter virus. NS1 proteins are highly conserved glycoproteins that regulate replication, spread, and neurovirulence. Mutating any one of three NS1 N-linked glycosylation sites (N85, N207/208, N223/224) reduced replication kinetics and attenuated replication by 1-2 logs reduced NS1 secretion and delayed cell-to-cell spread. Mutating the N224Q glycosylation site resulted in a highly attenuated recLI9 mutant severely restricted replication and foci formation in vitro, and in vivo enhanced survival of inoculated mice consistent with reduced POWV neuroinvasion.
Passage Attenuation of POWV L19-P:The most successful FV vaccine (YFV-17D) was generated by passage attenuation of virulent YFV-Asibi in cell culture. Using a similar approach, we generated passage attenuated LI9-P, that contains 8 residue changes in proteins that mediate cell attachment and IFN regulation. LI9-P replicates like LI9 (titer/kinetics) in vitro and forms infected cell foci, however, LI9-P fails to enter the CNS, cause weight loss or lethality in mice. LI9-P elicits neutralizing POWV antibodies, protects mice from LI9 challenge and is a candidate live attenuated POWV vaccine. In contrast to LI9, LI9-P highly induces IFN beta/lambda responses in BMECs and CPEpCs. Our findings are consistent with residue changes in LI9-P Env and NS3/4A/4B proteins restricting neuroinvasion and provide reverse genetics analysis targets. Contrasting LI9 and LI9-P spread at neurovascular barriers, permits us to resolve mechanisms of LI9 neuroinvasion, in a lethal POWV murine model.
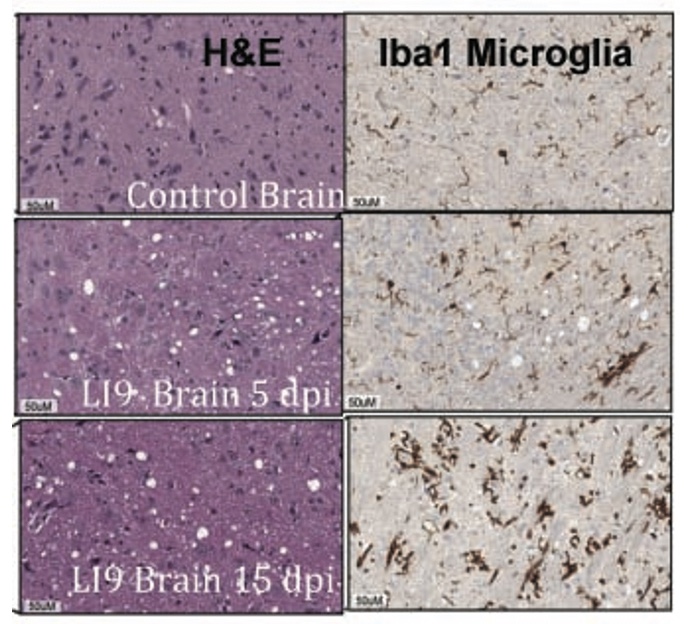
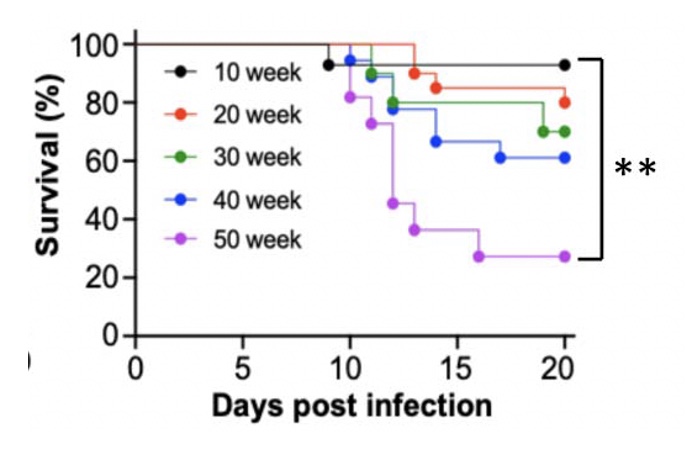
Age-Dependent Lethality of POWV L19: B6 mice footpad inoculated with LI9 develop lethal neurovirulent disease with overt spongiform CNS damage and microglial activation. LI9 is 82% lethal in 50 wk old mice with lethality sequentially reduced in 40/30/20/10 wk old mice to 7.1%. Brains of LI9 infected 50 wk old mice display neuronal loss and spongiform neuropathology by 5 dpi. Despite early damage, age-dependent lethality in 50 wk old mice occurs 10-15 dpi concomitant with prolonged CNS viral load and M1 type cytokine induction. Lethality and spongiform damage in LI9 infected aged mice is consistent with severe POWV disease in elderly patients, and spongiform CNS damage, inflammation and age directed “cell senescence” found in Alzheimer’s disease (AD). Zika Virus Persistent ZV Infections of Endothelial Cells. Zika virus (ZIKV) is a neurovirulent flavivirus that uniquely causes fetal microcephaly, is sexually transmitted, and persists in patients for up to 6 months. https://renaissance.stonybrookmedicine.edu/sites/default/files/1.jpegWe found that ZIKV (strain PRVABC59) persistently infects and continuously replicates in primary human brain microvascular ECs (hBMECs), without cytopathology, for >9 days and following hBMEC passage. ZIKV does not permeabilize hBMECs but was released basolaterally from polarized hBMECs, suggesting a direct mechanism for ZIKV to cross the BBB. ZIKV-infected hBMECs are rapidly resistant to interferon (IFN-a) and transiently induced, but failed to secrete, IFN beta or IFN lambda. We found that CCL5, a prosurvival chemokine, is highly secreted by ZIKV-infected hBMECs. The presence of CCL5 receptors (CCR3 and CCR5) on ECs suggest that CCL5 promotes ZIKV persistence in hBMECs. Induced CCL5 Fosters ZV Persistence. We found that exogenous CCL5 induced ERK1/2 phosphorylation in hBMECs and that ERK1/2 cell survival signaling was similarly activated by ZIKV infection. While KO of CCL5 failed to prevent ZIKV infection of hBMECs, we observed a 90% reduction in ZIKV-infected CCL5-KO hBMECs and a multilog reduction in ZIKV titers. In contrast, the addition of CCL5 to CCL5-KO hBMECs dose-dependently rescued ZIKV persistence in hBMECs. Inhibiting CCL5 responses using CCR3 (UCB35625) and CCR5 (maraviroc) receptor antagonists reduced the number of ZIKV-infected hBMECs and ZIKV titers. Thus ZIKV-induced CCL5 directs autocrine CCR3/CCR5 directed ERK1/2 survival responses that are required for ZIKV persistence in hBMECs. This establishes a role for CCL5 in ZIKV persistence and CCL5 receptor antagonists as potential therapeutics for limiting ZIKV spread and neurovirulence. ZV Post-Transcriptionally Regulates IFN Expression: We found that ZIKV induces IFN beta and IFN lambda mRNAs in hBMECs, but post-transcriptionally inhibits IFN beta/IFN lambda expression. IFN beta/IFN lambda mRNAs contain AU-rich elements (AREs) in their 3 untranslated regions which regulate protein expression through interactions with ARE-binding proteins (ARE-BPs). We found that ZIKV infection of primary hBMECs induces the expression of the ARE-BP tristetraprolin (TTP) and TTP is a novel regulator of endothelial IFN secretion. In hBMECs, TTP knockout (KO) increased IFN beta and IFN lambda abundance and IFN beta/IFN lambda secretion in response to ZIKV infection and inhibited viral persistence. In contrast, TTP expression dramatically reduced IFN beta/IFN lambda secretion in hBMECs. IFN beta/IFN lambda mRNA stability was not altered by TTP. TTP is similarly induced by ZIKV infection of Sertoli cells, and like hBMECs, TTP expression or KO respectively inhibited or enhanced IFN beta/IFN lambda mRNA levels in Sertoli cells. Thus ZIKV-induced TTP promotes viral persistence in hBMECs and Sertoli cells by post-transcriptioanlly regulating IFN beta/IFNλ lambda secretion. Our findings demonstrate a novel role for virally induced TTP in regulating IFN secretion in barrier cells that normally restrict viral persistence and spread to protected compartments. Dengue Virus - Role of Endothelial Cells in Dengue Infections: Dengue viruses (DVs) cause 2 vascular leak syndromes: dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). Initial DV infections do not cause DHF or DSS in patients or permeabilize the vasculature. However, a second DV infection, by a different DV serotype, gives rise to an immune enhanced disease process resulting in hemorrhage or edema in a small percentage of patients. Ultimately vascular permeability results from changes in the vascular endothelium. We found that primary human ECs are efficiently and productively infected by DV, and that DV infected ECs rapidly produce virus. DV regulates early but not late IFN responses, with late IFN beta induction directing viral clearance from ECs. On Transwell plates DV infection of polarized EC monolayers does not cause monolayer permeability by TEER or FITC dextran assays. DV infected ECs secrete enormous amounts of NS1 protein into infected EC supernatants without permeability, and the addition of NS1 containing supernatants to EC monolayers also fails to permeabilize ECs. These findings demonstrate that high level NS1 secretion from infected ECs does not cause of permeability in infected or uninfected ECs. This easily reproduced data contradicts studies suggesting that the addition of small amounts of recombinant NS1 added to ECs is a cause of monolayer permeability. DV-infected ECs secrete cytokines and chemokines that have the potential to contribute to immune enhanced pathogenesis. We are interested in determining the contribution of ECs to antibody dependent enhancement (ADE) of DV pathogenesis. We are also interested in the mechanisms by which DV proteins inhibit early but not late IFN signaling pathways and the role of DV directed EC cytokine responses in ADE. Studies include defining viral and cellular targets for therapeutically regulating DV directed EC responses that contribute to DV disease severity. SARS-C0V-2 Fails to Infect Vascular Endothelial Cells Since ECs Lack ACE2 Receptors: SARS-CoV-2 causes COVID-19, an acute respiratory distress syndrome (ARDS) characterized by pulmonary edema, viral pneumonia, multiorgan dysfunction, coagulopathy, and inflammation. SARS-CoV2 uses angiotensin-converting enzyme 2 (ACE2) receptors to infect and damage ciliated epithelial cells in the upper respiratory tract. How SARS-CoV-2 dysregulates vascular functions to cause ARDS in COVID-19 patients remains an enigma focused on dysregulated EC responses. We found that primary human ECs lack ACE2 receptors and that SARS-CoV2 is incapable of directly infecting ECs derived from pulmonary, cardiac, brain, umbilical vein, or kidney tissues. In contrast, when we transduced primary pulmonary ECs to express recombinant ACE2 receptors, we found that SARS-CoV2 lytically infected ECs, directing the production of high viral titers and multinucleate syncytia. The inability of SARS-CoV-2 to directly infect ECs without ACE2 expression explains a lack of EC lysis and vascular hemorrhage in COVID-19 patients, and indicates that the endothelium is not a primary target of SARS-CoV-2 infection. Our findings are consistent with SARS-CoV-2 indirectly activating EC programs that regulate thrombosis and endotheliitis in COVID-19 patients and focus therapeutic strategies that target epithelial and inflammatory responses that activate the endothelium. Hantavirus Pathogenic Mechanisms Pathogenic hantaviruses (HVs) primarily infect endothelial cells (ECs) and cause two vascular leak syndromes, hantavirus pulmonary syndrome (HPS) and hemorrhagic fever with renal syndrome (HFRS). Both diseases are characterized by acute thrombocytopenia with HPS resulting in an influx of up to 1 liter/hr of fluid in the lung. Beta 3 Integrin- and VEGF-Directed Permeability: Human beta 3 integrins play prominent roles in regulating vascular integrity through beta 3 directed platelet and EC adherence functions, with genetic mutations in beta 3 causing vascular permeability based bleeding disorders (Glanzmann’s Thrombasthenia). On ECs, beta 3 integrins temper the permeabilizing effects of vascular endothelial growth factor (VEGF) by binding to VEGF receptor 2 (VEGFR2). Beta 3 integrins are highly expressed on platelets and mediate their activation, deposition and clotting activity. We determined that HPS and HFRS causing HVs bind PSI domains on bent inactive beta 3 integrin conformers and enhance EC responses to the permeability factor VEGF. In contrast, the nonpathogenic HV, TULV, failed to bind beta 3 integrins or enhance VEGF permeability responses. We found that infection of primary human ECs with pathogenic (HFRS or HPS) causing HVs, but not TULV, resulted in the selective adherence of platelets to ECs, suggesting a mechanism of platelet sequestration during pathogenic HV infections. Analysis of HPS patient edema fluid found that VEGF is increased in HPS patients pulmonary fluids, potentially from hypoxia induced VEGF that causes high-altitude pulmonary edema and suggesting that an auto-amplifying VEGF-HIF1alpha loop likely contributes to the rapid fluid influx observed in HPS patients. RhoA/RhoGDI Regulation of EC Permeability: Andes virus (ANDV) nonlytically infects pulmonary microvascular ECs (PMECs), causing acute pulmonary edema and HPS. In HPS patients virtually every PMEC is infected, and in primary PMECs the ANDV nucleocapsid (N) protein activates the RhoA GTPase causing VE-cadherin internalization from inter-EC adherens junctions and PMEC permeability. The ANDV N protein fails to bind RhoA, however N protein coprecipitates the primary RhoA repressor, RhoGDI (Rho GDP dissociation inhibitor), that normally sequesters RhoA in an inactive state. Collectively, ANDV N binds C-terminal residues of RhoGDI, activating RhoA by sequestering suppressive RhoGDI. Infection of Pericytes: Pericytes are stromal cells, intermittently attached to the basolateral side of capillary microvascular ECs, that maintain homeostasis of the vascular barriers. We found that in vitro, ANDV persistently infects primary human vascular pericytes for up to 9 days, and that PMEC monolayer permeability was increased by supernatants from ANDV-infected pericytes. Pericyte-directed PMEC permeability was consistent with the high-level secretion of the permeability factor VEGF (vascular endothelial growth factor) elicited by ANDV-infected pericytes. These findings suggest that ANDV infection of pericytes increases PMEC permeability and reveals a novel mechanism of pericyte-directed vascular barrier dysfunction that contributes to HPS. 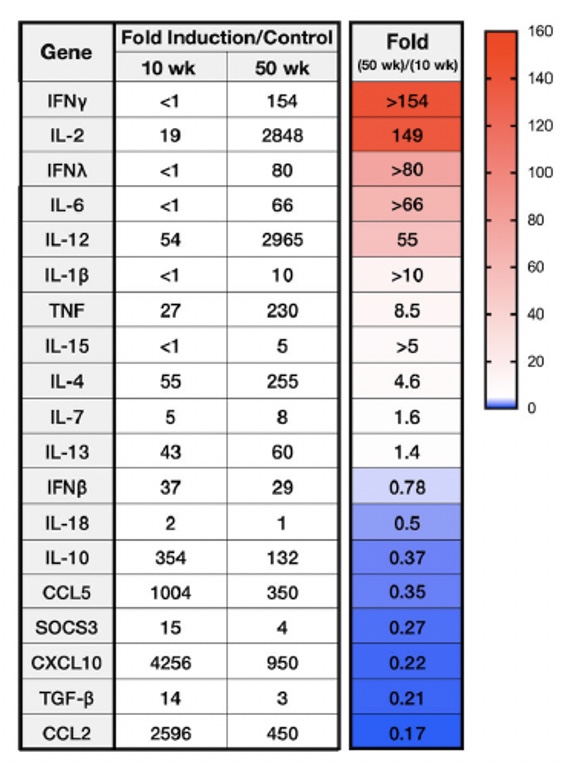 |
|
||
| Co-Investigators
Dr. Hwan Keun Kim, Department of Microbiology and Immunology, Stony Brook University Dr. Stella Tsirka, Department of Pharmacological Sciences, Stony Brook University |
|||
| Senior Research Associates and Postdoctoral Fellows
Elena Gorbunova, Ph.D. Megan Mladinich Valenti, PhD. |
|||
|
Graduate Students Grace Himmler, Molecular and Cell Biology PhD Program Marissa Lindner, Microbiology and Immunology PhD Program |
|||
|
Laboratory Associates Huiyeon Kim Gabriela Riera
|
|||
| Publications
Selected Publications Mladinich, M, Himmler, G, Conde, J., Gorbunova, E. , Schutt, W.R., Tsirka, S., Kim, H.K., and Mackow, E.R. 2024. Age-dependent Powassan Virus Lethality is Linked to Glial Cell Activation and Divergent Neuroinflammatory Cytokine Responses in a Murine Model. J. Virol., in press. Schutt, W.R., Conde, J.N., Mladinich, M.C., Himmler, G.E., and Mackow, E.R. 2023. ZIKV Induction of Tristetraprolin in Endothelial and Sertoli Cells Post-Transcriptionally Inhibits IFNβ/λ Expression and Promotes ZIKV Persistence. mBio 14(5). DOI: https://doi.org/10.1128/mbio.01742-23 Conde, J., Himmler, G, Mladinich, M., Setoh, YX., Amarilla, A.A., Schutt, W., Saladino, N., Gorbunova, E., Salamango, D., Benach, J., Kim, H.K., Mackow, E.R. 2023. Establishment of a CPER Reverse Genetics System for Powassan Virus Defines Attenuating NS1 Glycosylation Sites and an Infectious NS1-GFP11 Reporter Virus. mBio 10.1128/mbio.01388-23. Conde, J.N., Sanchez-Vicente, S., Saladino, N., Gorbunova, E.E., Schutt, W.R., Mladinich, M.C., Himmler, G.E., Benach, J., Kim, H.K., Mackow, E.R. 2022. Powassan Viruses Spread Cell to Cell during Direct Isolation from Ixodes Ticks and Persistently Infect Human Brain Endothelial Cells and Pericytes. J. Virology. 12;96(1):e0168221 [PMCID: PMC8754205]. Sohn, S.Y., Hearing, J., Mugavero, J., Kirillov, V., Gorbunova, E., Helminiak, L. Mishra, S., Mackow, E, Hearing, P., Reich, N. and Kim, H.K. 2021.Interferon-lambda Protection and Differential Sex Pathology in a Murine Model of SARS-CoV-2 Infection. mBio 12(6): e02756-21. 10.1128/mBio.02756-21. Conde, J.N., Sanchez-Vicente, S., Saladino, N., Gorbunova, E., Schutt, W.R., Mladinich, M., Himmler, G.E., Benach, J., Kim, H.K., and Mackow, E.R. 2022. A Novel Powassan virus isolate is Basolaterally Released from Persistently Infected Primary Human Brain Endothelial Cells and Pericytes. J. Virol. 96(1): e0168221. Perez, R., Gorbunova, E., and Mackow, E.R. 2021. Novel Infection of Pericytes by Andes Virus Enhances Endothelial Cell Permeability. Virus Research. 306 198584. 10/2021. https://doi.org/10.1016/j.virusres.2021.198584. Mladinich, M., Conde, J.N., Schutt, W.R., Sohn, S.Y., and Mackow, E.R. 2021. Blockade of Autocrine CCL5 Responses Inhibits Zika Virus Persistence and Spread in Human Brain Microvascular Endothelial Cells. mBio 12:e01962-21. Gorbunova, E., and Mackow, E. 2021. Binding of the Andes Virus Nucleocapsid Protein to RhoGDI Induces the Release and Activation of the Permeability Factor RhoA. Journal of Virology 95:e00396-21. https://doi.org/10.1128/JVI.00396-21. Conde, J.N., Schutt, W., Gorbunova, E.E., and Mackow, E.R. 2024. Recombinant ACE2 Expression is Required for SARS-CoV-2 to Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 11/2020 in press. https://biorxiv.org/cgi/content/short/2020.11.10.377606v1 Conde, J.N., Schutt, W., Mladinich, M., Sohn, S.Y., Hearing, P., and Mackow, E.R. 2020. NS5 Sumoylation Directs Nuclear Responses that Permit Zika Virus to Persistently Infect Human Brain Microvascular Endothelial Cells. J Virology doi:10.1128/JVI.01086-20. Simons, M.J., Gorbunova, E.E., Mackow, E.R. 2019. Unique Interferon Pathway Regulation by the Andes Virus Nucleocapsid Protein Is Conferred by Phosphorylation of Serine 386. J. Virology 93:e00338-19. Mladinich, M., Schwedes, J., Mackow, E.R. 2017. Zika Virus Persistently Infects and Is Basolaterally Released from Primary Human Brain Microvascular Endothelial Cells. mBio 8, 4 e00952-17. |
|||