This chapter is intended as an introduction to the basic concepts of the pathophysiology of ischemic injury of the brain and cerebrovascular disease.
Specific topics:
The general features of energy metabolism of the brain, the concept of selective vulnerability and the concept of excitotoxicity
Definition of stroke
The clinical presentation of stroke
The types of strokes and the major causes of each
The relationship between vascular anatomy and the cause of stroke
The evolution of reactive changes in the brain following a stroke General features of energy metabolism of the brain, selective vulnerability and excitotoxicity
Delivery of oxygen and glucose to the brain
Under normal circumstances, the brain uses only glucose for fuel. Oxygen is needed for its metabolism. Ventilation and circulation are required to perfuse the brain. This means that dysfunction of other systems, such as the cardiovascular and pulmonary systems can result in brain injury.
The perfusion pressure of the brain is defined by the following relationship:
Cerebral perfusion pressure = mean arterial pressure -intracranial pressure
One consequence of this relationship is that as intracranial pressure increases, cerebral perfusion decreases. Increased intracranial pressure can be due to many processes, including brain swelling due to either cytotoxic or vasogenic edema, or any space-occupying lesion such as a hematoma, tumor or abscess.
Within certain limits, regardless of mean arterial pressure, cerebral blood flow is maintained at a constant level. This is known as cerebral autoregulation. The average cerebral blood flow (CBF) is 50ml/100gm/minute, but gray matter has greater blood flow than white matter. This means that, in most circumstances, gray matter is more vulnerable to interruption of blood flow, for whatever reason, than white matter.
Local CBF is coupled to metabolism so that brain areas that are electrically active have more blood flow than areas that are relatively quiescent and have lower electrical and metabolic activity. This is known as local autoregulation. The factors responsible for this process are not known, but might include pH, adenosine concentration or nitric oxide.
Brain energy metabolism
Glucose is the main source of fuel in the brain. It is metabolized through the glycolytic pathway to pyruvate and then through the tricarboxylic acid cycle (TCA). The NADH and FADH2 that are formed are then are used for oxidative phosphorylation. (Some glucose is metabolized through the Pentose Phosphate shunt- forms NADPH). Glia can also convert pyruvate into oxaloacetate to enter TCA cycle. The TCA cycle is a source of gamma-aminobutyric acid (GABA), from a-ketoglutarate and also a source of aspartate, which is formed from oxaloacetate.
Ischemia- Ischemia is lack of blood flow.
Global ischemia can affect the entire brain. It is usually due to a process occurring outside the brain, for example, in cardiac dysfunction. Focal ischemia affects only a limited geographic region of brain. Ischemia can cause necrosis, either selective necrosis of neurons only or infarction, which is death of all of the elements in the region of tissue. Neurons are more vulnerable to ischemia than glia.
Effects of ischemia-
Glucose and Oxygen are not delivered. Metabolic waste is not removed. Nitric oxide, produced by neurons, glial and inflammatory cells responding to ischemic injury, contributes to neuronal damage by acting as a free radical. Excessive production of glutamate and/or aspartate can result in excitotoxicity.
The effects of ischemia are modulated by a number of factors, including: duration of ischemia- Under usual conditions, neurons can survive 4 minutes when perfusion ceases; consciousness is lost in 10 seconds.
degree of ischemia
brain temperature - Hypothermia is protective (decreases metabolic demands, decreases free radicals).
blood glucose level - Hypoglycemia is protective, while hyperglycemia results in excess lactate and acidosis. Experiments in which glycolysis is blocked show decreased damage from ischemia. |
Selective neuronal necrosis is due, at least in part, to excitotoxicity, which occurs when glutamate, released in ischemia, causes overwhelming influx of calcium into dendrites, resulting in neuronal death. In experimental settings, glutamate receptor antagonists have been shown to block the neuronal necrosis that can occur as a result of ischemia. In Vitro studies have demonstrated that synaptic activity is necessary for hypoxic neuronal necrosis, providing further evidence that it is synaptic activity resulting in excitotoxicity that is responsible for this neuronal death.
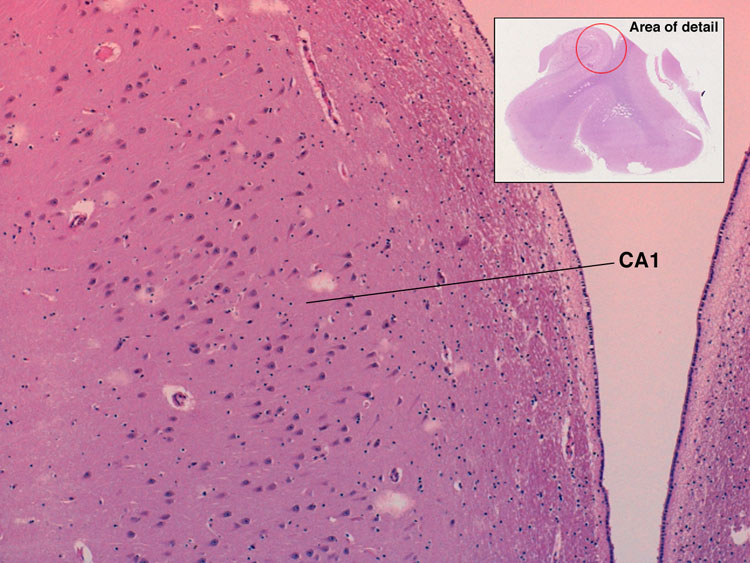
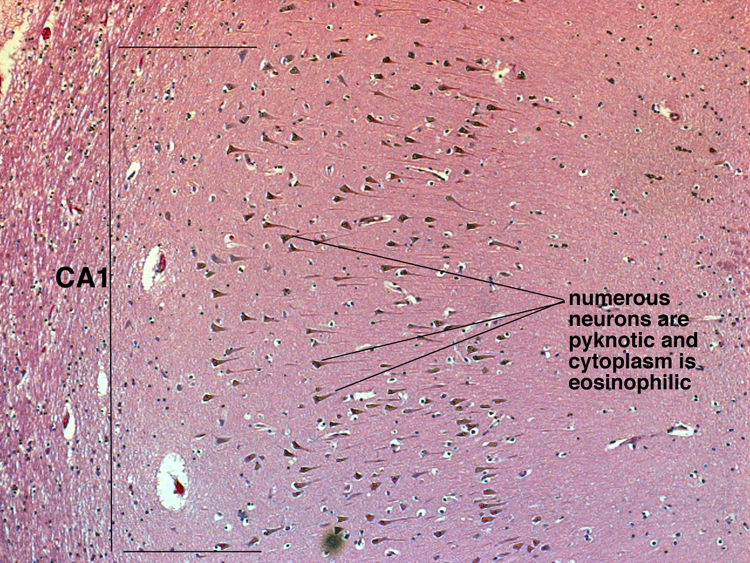
| There are also regions of brain that are known to be more vulnerable to global hypoperfusion. These include: CA1 region of the hippocampal formation, Purkinje cells of the cerebellum, the Globus Pallidus and layers 3 and 5 of the cerebral cortex. It appears that this vulnerability is due to specific properties of the neurons in those zones, probably a consequence of their neurotransmitter receptor subtypes. |  |
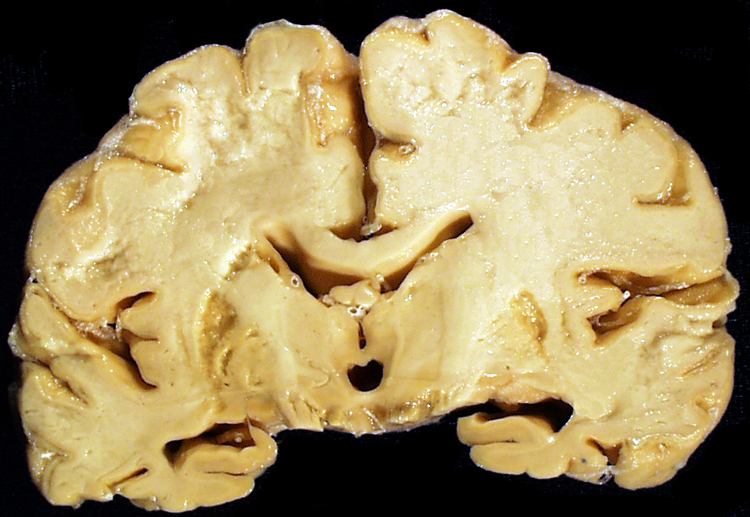
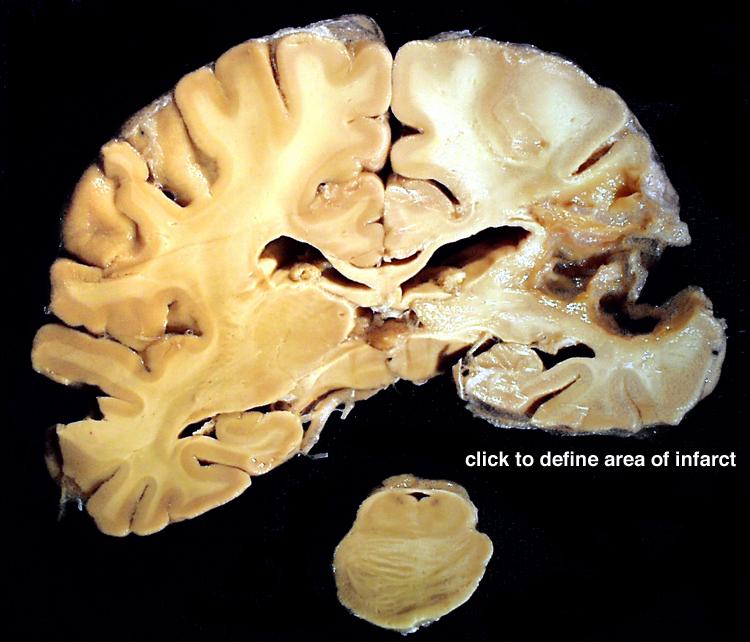
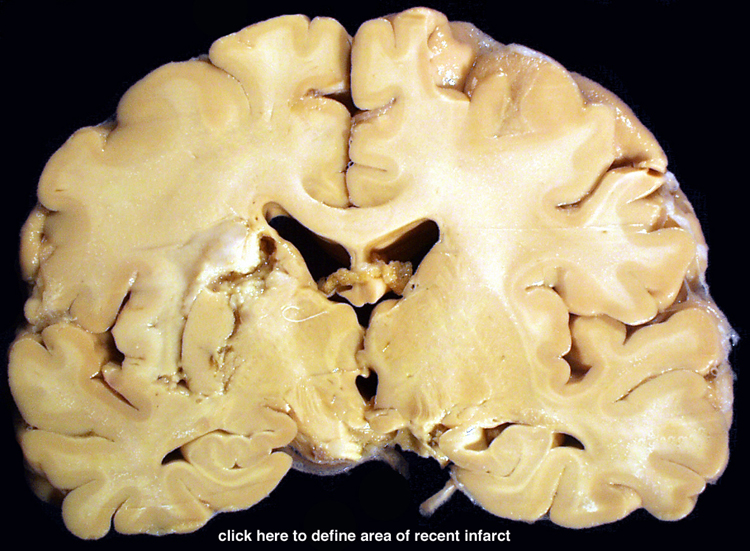
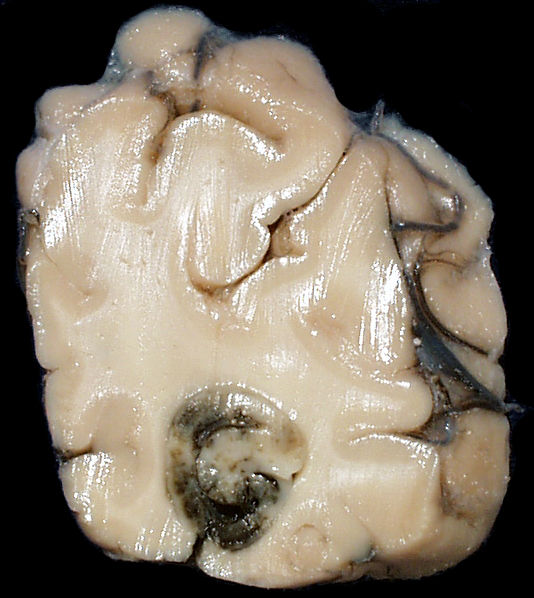
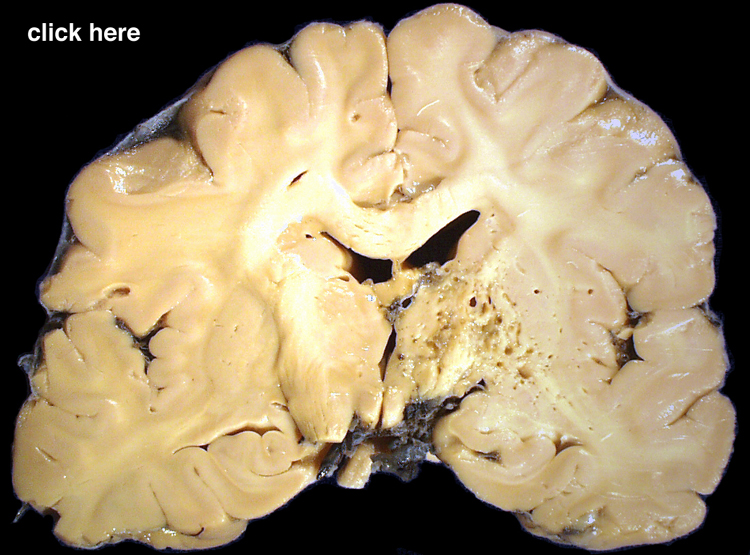
| This coronal section of the cerebral hemispheres demonstrates the result of a severe global hypoxic-ischemic insult. It is from a person who was resuscitated after experiencing a large pulmonary embolus with severe hypoxia and hypotension. She lived several months following the event, first in coma, then in a vegetative state. This section shows marked thinning of the majority of the cortical ribbon (compare to normal) and atrophy of the deep gray structures. The lateral ventricles are secondarily enlarged; this passive enlargement of the ventricles as a result of loss of brain tissue is known as hydrocephalus ex vacuo. | 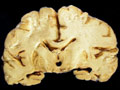 |
Stroke-
Definition" rapidly developing clinical signs of focal (at times global) disturbance of cerebral function, lasting more than 24 hours or leading to death with no apparent cause other than that of vascular origin" (World Health Organization)
Clinical presentation - A sudden onset of symptoms is characteristic of vascular events, however, it is important to note that other disorders can present suddenly. In stroke, the initial neurological signs and symptoms reflect the functional anatomy of the region of the brain affected , but later, secondary changes can occur, which will alter the clinical signs. These later changes can include herniation due to swelling or vasospasm, particularly in the presence of blood. Vasospasm can cause infarction of additional brain tissue.
Risk factors
Untreatable-increasing age, male, African American, heredity
Treatable - hypertension, diabetes mellitus, cigarette smoking, obesity, hypercholesterolemia, heart disease, history of prior transient ischemic attack (TIA)
General concepts about vascular anatomy and its relationship to the pathophysiology of stroke:
Superficial vessels are most commonly occluded by emboli. Deep penetrating vessels are most commonly affected by hypertension. Watershed zones are the border zones between the distal locations of the territories supplied by two arteries and are vulnerable to hypotension. One can review the territories of supply of the major arteries of the brain in any standard anatomy text.
Focal ischemic infarction-
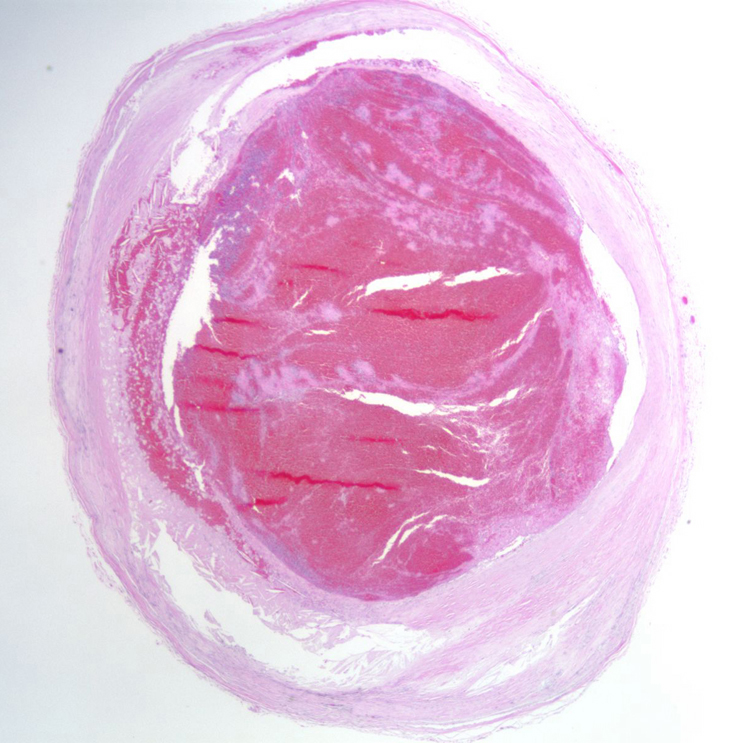
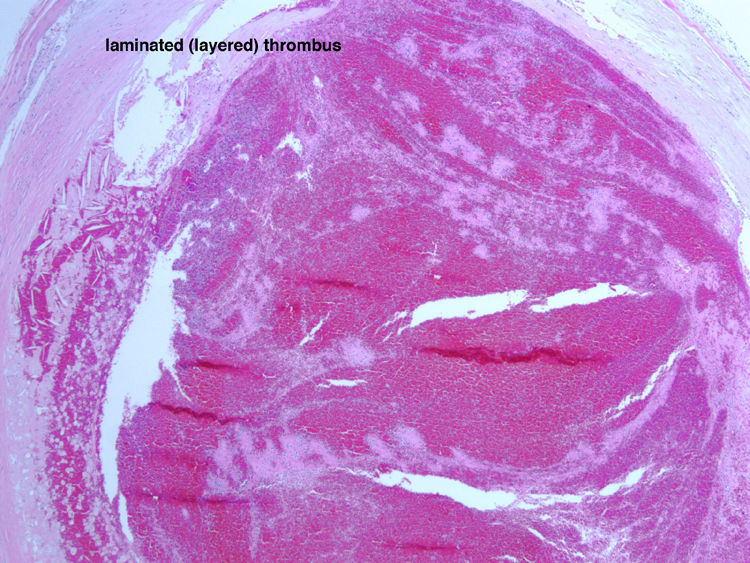
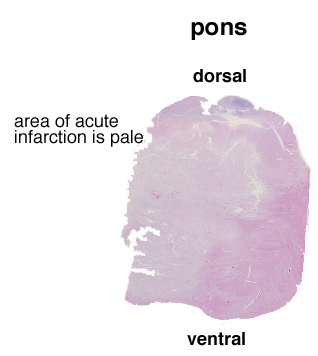
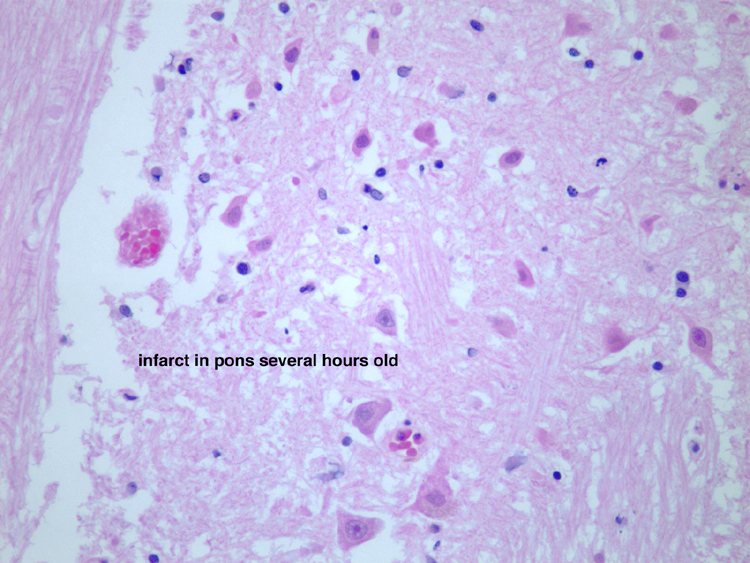
Infarction is death of tissue in a region of brain due to lack of blood flow through the blood vessel that supplies that territory. The main pathophysiological processes that can result in focal ischemic infarction are: atherosclerosis, embolism, arteriosclerosis and hypotension. | Atherosclerosis- atherosclerotic plaques can directly occlude blood vessels by progressive stenosis (narrowing) of the lumen of the affected blood vessel. Sometimes, hemorrhage within an atherosclerotic plaque can cause obliterate the lumen. Atherosclerosis can also serve as a nidus for thrombosis, with endothelial surface abnormalities serving as a site for activation of the clotting cascade. Atherosclerotic plaques can break off and embolize to downstream vessels (see embolism, below). A section demonstrates acute basilar artery thrombosis which developed at the site of atherosclerosis, which provides a nidus for thrombosis. At higher power, the cholesterol clefts of the atherosclerotic plaquelaminated, which indicates that it was formed when blood was flowing, rather than post-mortem. A section of the pons from this patient, an 86 year old woman with a history of hypertension, who had not been seen by relatives in a few days, shows a large area of pallor, which is acute infarction of the pons. A high power photomicrograph demonstrates the pontine eosinophilic neurons which are consistent with acute infarction several hours old. are visible. The thrombus is | |
Embolism- A variety of materials within the circulation can travel downstream and lodge in vessels with lumens too narrow to allow passage of the material. Small emboli tend to lodge in distal branches of superficial vessels which results in infarction at the gray-white junction. Multiple small infarcts distributed throughout the cerebral hemispheres at the gray-white junction are characteristic of embolic infarction. The occurrence of a single large wedge-shaped infarct is also another common pattern of cerebral embolism. The most common location of embolic infarction is in the territory of the middle cerebral artery ("the artery of embolization"). Emboli can come from a variety of sources, including: artery-to-artery, the heart, air emboli due to surgical procedures and bone marrow (following long bone fracture).
There can be artery-to-artery embolization when fragments of atherosclerotic plaques are dislodged and embolize to distal blood vessels or when fragments of thrombi can embolize.
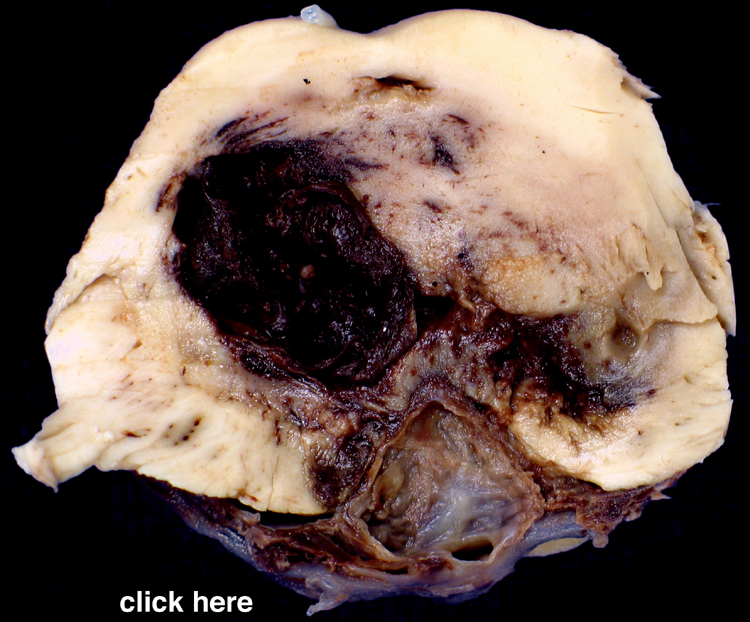
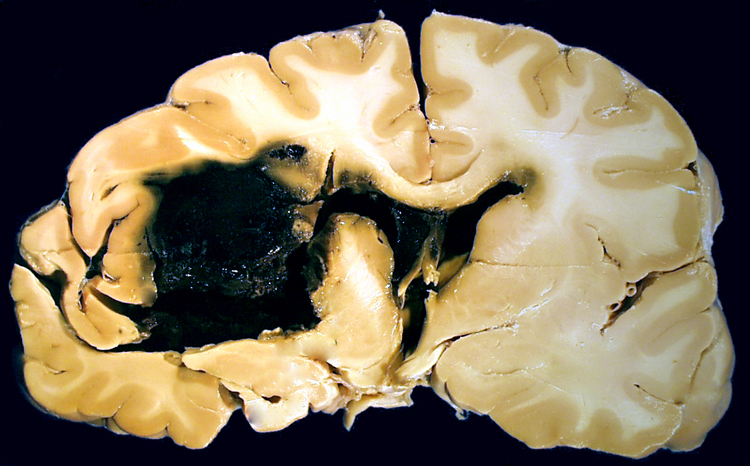
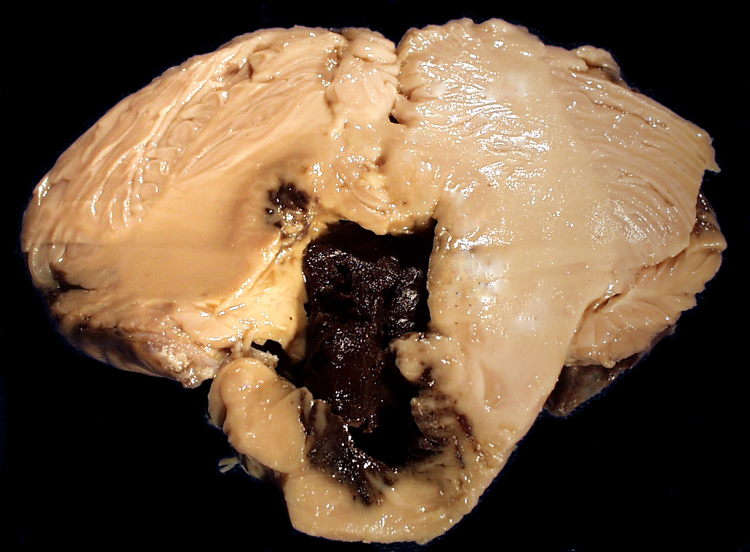
The heart is a common source of embolic material. Bacterial or non-bacterial vegetations on valves, atrial thrombi that develop in atrial fibrillation and endocardial mural thrombi following myocardial infarction are relatively common types of cardiac emboli. | A coronal section of cerebral hemispheres through the parietal lobes demonstrates a wedge-shaped area of expansion with brown stippling that represents recent hemorrhagic infarction most likely due to embolism to a branch of the middle cerebral artery (MCA). A section of another brain, at a slightly more posterior level demonstrates a wedge-shaped zone of parenchymal loss in the MCA territory. This represents remote hemorrhagic infarction, also probably due to embolism. A transverse section of pons is also included, which demonstrates atrophy of the pontine base on the same side as the hemispheric infarct. This is due to Wallerian degeneration of the corticospinal tract that originates in the ipsilateral cerebral cortex. | |
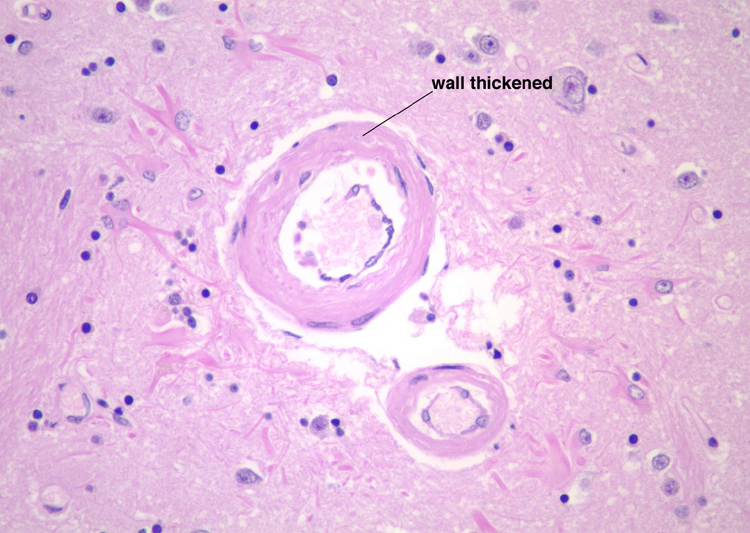
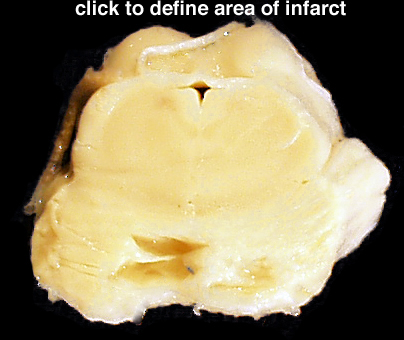
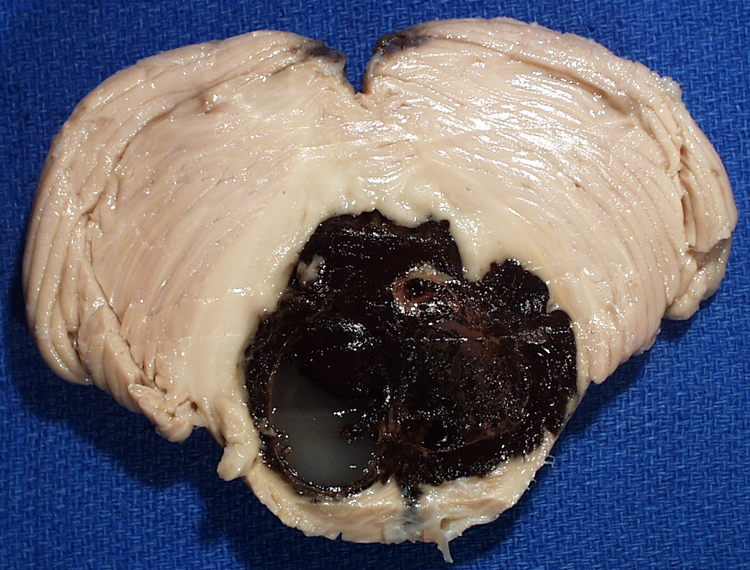
| Arteriosclerosis- This is disease of smaller arterial blood vessels that is most commonly due to hypertension. In the brain, arteriosclerosis due to hypertension affects the deep penetrating vessels that emerge at right angles to their parent vessels. Infarction due to hypertension is most commonly found in the basal ganglia, thalamus, pons and cerebellum. A photomicrograph of a section of basal ganglia demonstrates a stage of hypertensive arteriopathy in which there is thickening and hyalinization of the wall of this artery. A mild astrocytic gliosis is present in the surrounding parenchyma. A section of basal ganglia demonstrates a cavity in the putamen due to remote infarction. In this section of cerebral hemispheres, an area of expansion and early fragmentation (some of the fragmentation is technical in origin) represents a recent infarct in the basal ganglia. A section of pons demonstrates a remote infarct in the basis pontis due to hypertension. | |
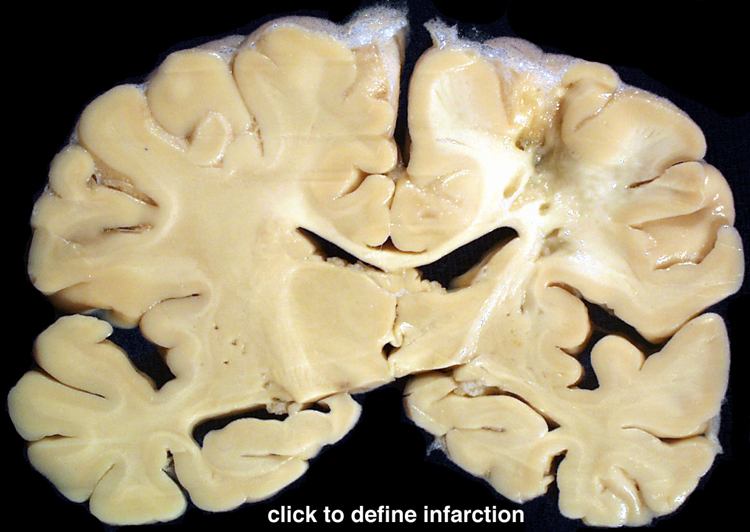
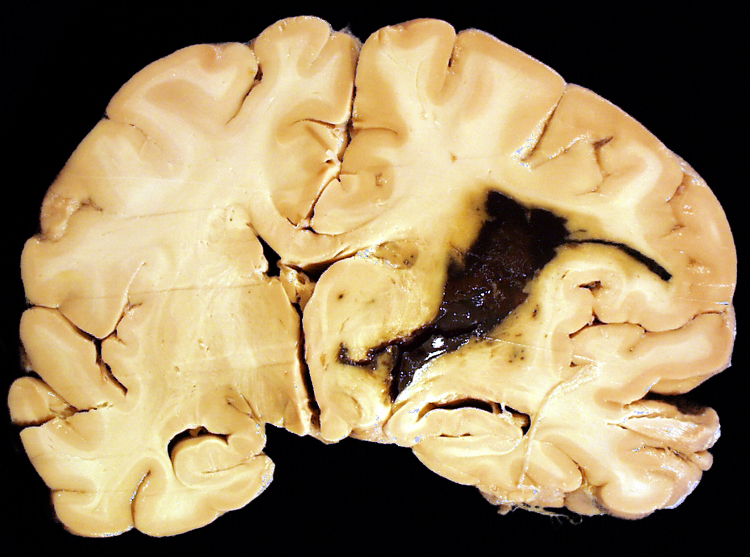
| Watershed infarction- This is infarction in the territory served by the overlap between the distal ends of two arteries. It is caused by generalized hypotension. Generalized or systemic hypotension can also cause other patterns of infarction (please see section V, above). A section of cerebral hemispheres demonstrates recent bilateral watershed infarction, in the territories supplied by the distal branches of the anterior and middle cerebral arteries. The areas of infarction are expanded due to the presence of edema and they are discolored with brown stippling due to hemorrhage into the infarcted zone. Another section of cerebrum demonstrates remote watershed infarction, in which there is an irregular cavity at the site of the infarct. | |
There are numerous other, less common causes of infarction of the brain. Hemorrhagic infarction-
Most physicians use this term without precision to refer to any cerebrovascular event in which there is hemorrhage, which includes both hemorrhage and hemorrhagic infarction. We will use the more accurate restricted concept of this term, which does not include primary CNS hemorrhage. Hemorrhagic infarction occurs when there is initially lack of blood flow through a vessel which causes infarction and then there is at least some reperfusion, which results in hemorrhage into the tissue that is infarcted (or already dead). Hemorrhagic infarction is commonly embolic in origin, presumably due to lysis of the embolus with restitution of blood flow. It also occurs sometimes in other situations when blood flow is reestablished to the devitalized tissue, such as following watershed infarction. It can also occur when blood flow is supplied by collateral anastomoses. A section of occipital lobe reveals a small acute hemorrhagic infarct, in this case, most likely due to a small embolus to a distal branch of the posterior cerebral artery. |  |
Evolution of the pathological process -
Following an ischemic event in the brain, gross and microscopic changes evolve in a predictable pattern. It is important to have an appreciation of the general time course of these changes for several reasons: the evolution of the pathological findings bears a relationship to the evolution of the clinical findings and can assist you in preparing the patient and family for what to expect, knowledge of what is occurring at the tissue level can direct therapy to maximize recovery and minimize secondary central nervous system damage as a result of the reactive changes, and sometimes the pathologist must determine if the changes in the brain correspond to the timeline of events, particularly in forensic pathology cases.
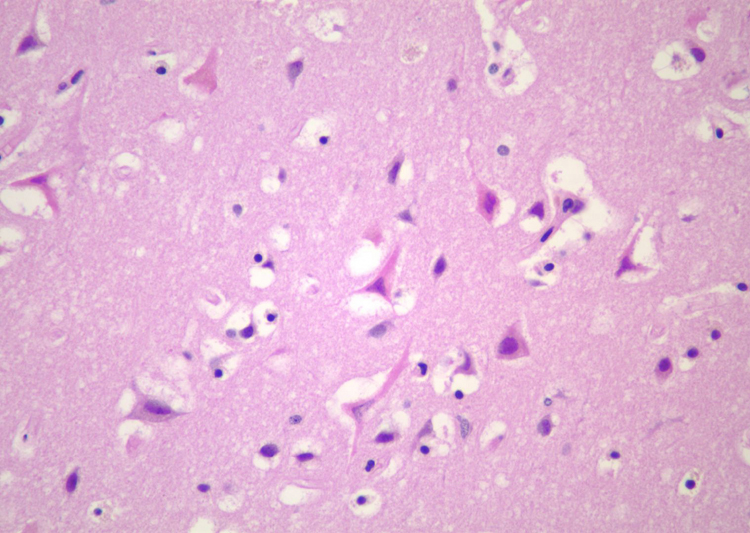
Gross changes - Initially, up to several hours following an infarct, there may be no detectable gross changes in the brain. After a few to several hours, there may be slight swelling and softening of the tissue and slight changes in the color of the affected tissue. The swelling and softening increase over the next several days. There is loss of distinction of the gray-white junction in cerebral cortex in the case of a cerebral cortical infarct. At about a week, the infarcted area begins to separate from the adjacent viable brain tissue. The next several weeks to months are characterized by decreasing swelling and increasing cavitation, until finally the infarcted area is a cavity. | Microscopic changes - Immediately after an infarct, up to approximately the first four to six hours, there may be no microscopic changes. The first detectable change is that the neurons in the area of infarction develop cytoplasmic eosinophilia (the cytoplasm looks pink-red in sections stained with hematoxylin and eosin). Neuronal shrinkage and perineuronal vacuolation develops. This section of cerebral cortex demonstrates acutely necrotic eosinophilic neurons and perineuronal vacuolation. | 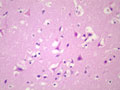 |
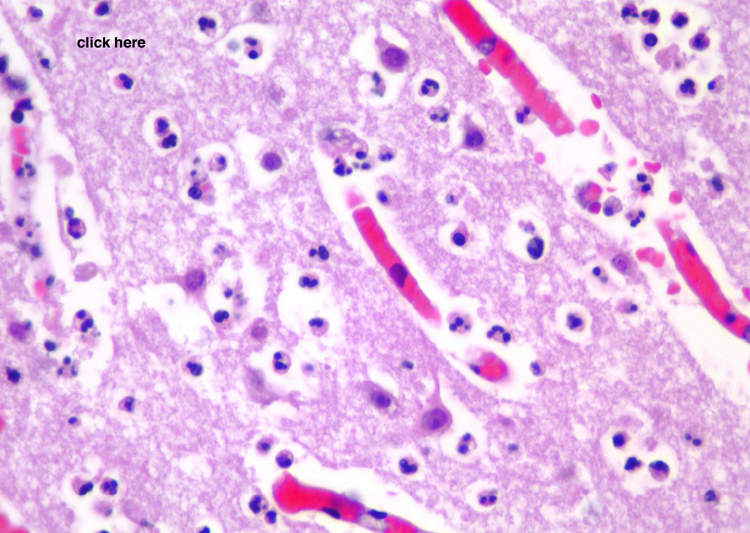
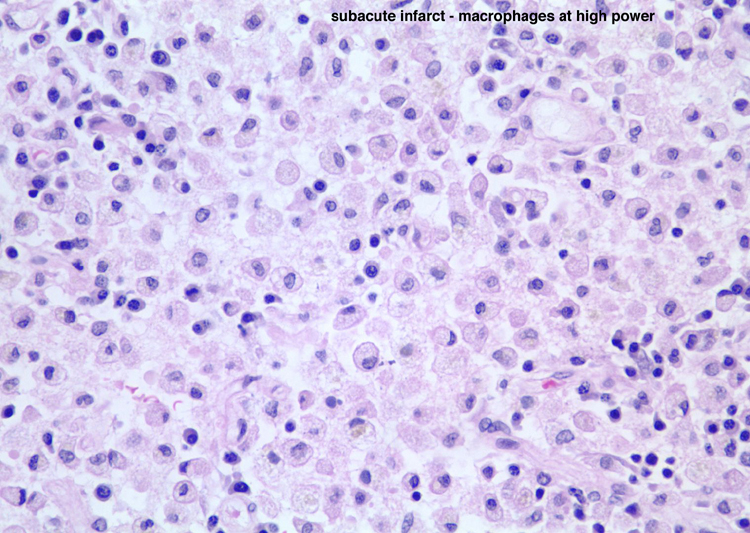
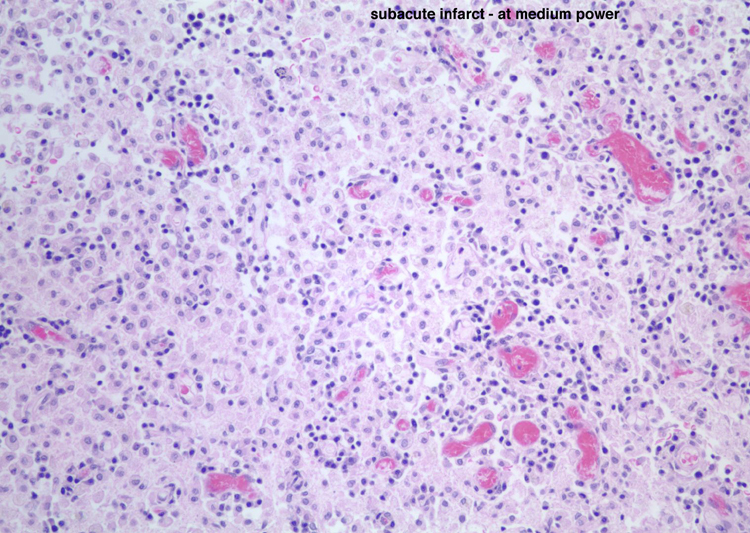
| Neutrophils enter the tissue and are the dominant reactive cell during the first 24 hours. This section demonstrates an infarct at approximately 24 hours with infiltration by neutrophils. Following this, macrophages enter the tissue and become the major reactive cell type from after the first day and continuing, gradually decreasing in density after the first several weeks, for weeks to months to even years, depending on the size of the infarct. Reactive astrocytes begin to be apparent from as early as the first day or so and they form the scar tissue as the process organizes over time. These sections demonstrate an ischemic infarct that is approximately two weeks old, in which there is a dense macrophage infiltrate in the territory of the infarct. | |
Intracranial hemorrhage -
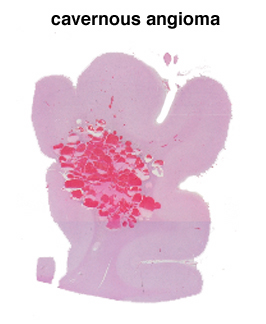
Subarachnoid hemorrhage is hemorrhage within the subarachnoid space, most commonly caused by rupture of a saccular (berry) aneurysm that develops in the vessels at the base of the brain. Subarachnoid hemorrhage presents as the sudden onset of the worst headache of one's life. The patients often have stiff neck, change in mental status, and subarachnoid hemorrhage on computed tomography (CT) of the brain. If the CT is normal and subarachnoid hemorrhage is still suspected, lumbar puncture can demonstrate blood in the cerebrospinal fluid. Approximately 2% of adults have saccular aneurysms and there is a risk of rupture of 23% per year. The greatest risk of rupture is from 40-60 years of age. 1/3 of patients with catastrophic subarachnoid hemorrhages are dead before treatment, 1/3 die in hospital or have serious sequelae and 1/3 do well. A very high percentage have warning signs which represent minor hemorrhages, most often in the two weeks prior to a catastrophic hemorrhage. It is important to recognize these "sentinel" hemorrhages, because the catastrophic hemorrhage can often be prevented. Other, less common causes of spontaneous (non-traumatic) brain hemorrhage include amyloid angiopathy, arteriovenous malformation, other vascular malformations, and bleeding disorders. Occasionally, a saccular aneurysm ruptures into the parenchyma resulting in parenchymal hemorrhage, rather than subarachnoid hemorrhage. Amyloid angiopathy can be suspected as the cause of a hemorrhage in an older individual who presents with lobar hemorrhage (a lobe of cerebrum) rather than in the basal ganglia. In the case of coagulopathy, there are often multiple, randomly distributed hemorrhages. | This section of cerebral hemispheres reveals a vascular malformation, probably an arteriovenous malformation within the deep gray structures. This section reveals the characteristic circumscribed appearance of a cavernous angioma within the subcortical white matter of the cerebrum. A higher power view demonstrates the thin-walled, back-to-back blood vessels that characterize this lesion. The patient with this cavernous angioma had a history of seizures. | |
Venous infarction
Hemorrhagic infarction due to vascular occlusion on the venous side of the circulation is relatively uncommon, but it does occur. This is most commonly due to thrombosis of a dural sinus. The most commonly affected are: superior sagittal sinus- 72%, lateral (transverse and sigmoid)-70% and the straight sinus -16%. It should be obvious that often there are multiple sinuses affected. True incidence of sinus thrombosis is not known and estimates of incidence vary from 16/12,500 autopsies to 9% of 182 consecutive autopsies. The mortality is estimated at about 10% and there Female/Male ratio is 1.29/1.
Predisposing factors for dural sinus thrombosis are as follows:
Infection- both systemic and CNS
Noninfective local factors: head injury, neurosurgery, cerebral infarction and hemorrhage, brain tumors
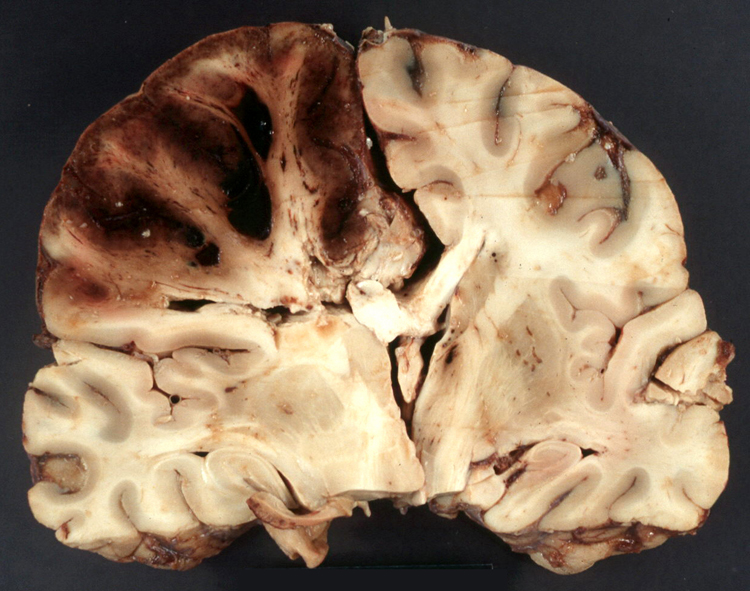
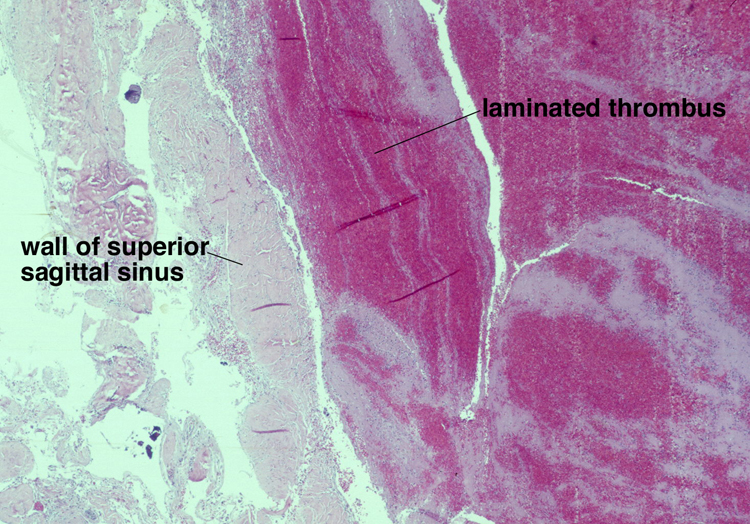
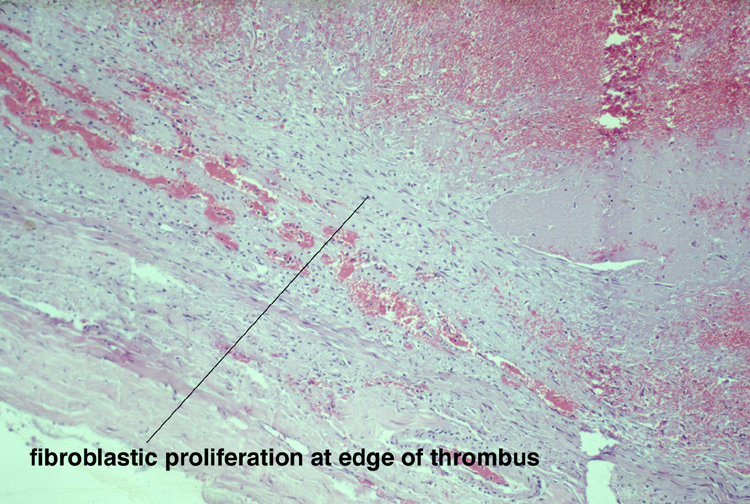
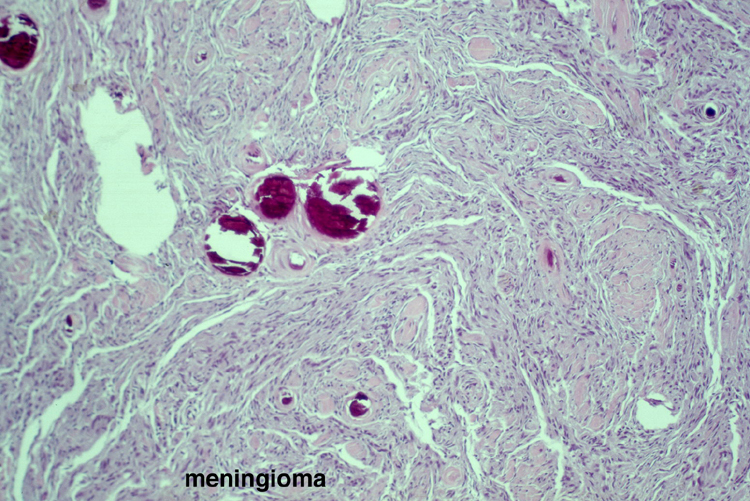
Noninfective general factors: Surgery (with or without deep venous thrombosis) pregnancy, puerperium, oral contraceptives, congenital cardiac disease, polycythemia, sickle cell disease, thrombocythemia, malignancy, coagulation disorders (circulating anticoagulants, DIC) dehydration, connective tissue diseases. The signs and symptoms of venous thrombosis include: headache (75%), papilledema (54%), focal motor or sensory deficit (34%), seizure (37%) and altered level of consciousness (30%). Neuroimaging reveals thrombosed veins, absence of enhancement of the affected sinus, increased enhancement of congested veins and dura and hemorrhagic infarcts. | This section of cerebral hemispheres demonstrates hemorrhagic venous infarction due to superior sagittal sinus thrombosis. A section of brain parenchyma demonstrates markedly distended congested veins and extravasation of blood. Sections of the superior sagittal sinus demonstrate laminated thrombus that is undergoing organization (fibroblastic proliferation). There was a small meningioma in the frontal region attached to the dura at the superior sagittal sinus. The extent of the organization indicates that this process had been going on for at least several days, probably longer. These sections are from a 48 year old woman with a chronic seizure disorder who presented with a few hours of confusion and lethargy. Within hours after admission to the hospital she lapsed into coma and died the following day. | |
|