Meet the research personnel in the Piret Lab
Lab News
RESEARCH
Our overall aim is to understand the molecular and cellular changes that occur in kidney proximal tubules in acute kidney injury (AKI) and progression of AKI to chronic kidney disease (CKD).
We have two main areas of focus: (1) the role of cellular metabolism in normal kidney, and its disturbances in AKI and transition to CKD; and (2) the role of RNA polymerase II in kidney responses to DNA damage in AKI and CKD.
Why is this important?
- In 2014, there were almost 4 million hospitalizations with AKI in people aged 20 and older
- Of patients aged 66 and older who were hospitalized with AKI in 2018, >8% died in hospital. Of those that were discharged, the cumulative 1-year incidence of recurrent hospitalization with AKI was >26%, and of death was >31%. Of those who did not have CKD before AKI, >50% went on to develop CKD after 1 year
- Of patients aged 20 and older that started outpatient dialysis for AKI treatment in 2018, the cumulative incidence of death at 1 year was >34%
- Patients in the ICU may have an incidence of AKI ranging from 20%–50%
- There are currently no therapies to treat AKI, reverse its effects, or prevent transition to CKD
Why the proximal tubule?
- The proximal tubule is the powerhouse for reabsorption of solutes and proteins by the kidney
- AKI may result from diverse causes such as drugs (e.g. chemotherapeutics), environmental toxins, sepsis, ischemia (lack of oxygen). All of these primarily affect the proximal tubule.
- Many cellular changes occur in damaged proximal tubules, including changes in morphology, cell cycle, secretome, and cellular metabolism
- We have shown that proximal tubular KLFs such as KLF6 and KLF15 are critical for determining the cellular response to injury, through regulating cellular metabolism and DNA damage responses. Our current research aims to understand the mechanisms underlying these effects
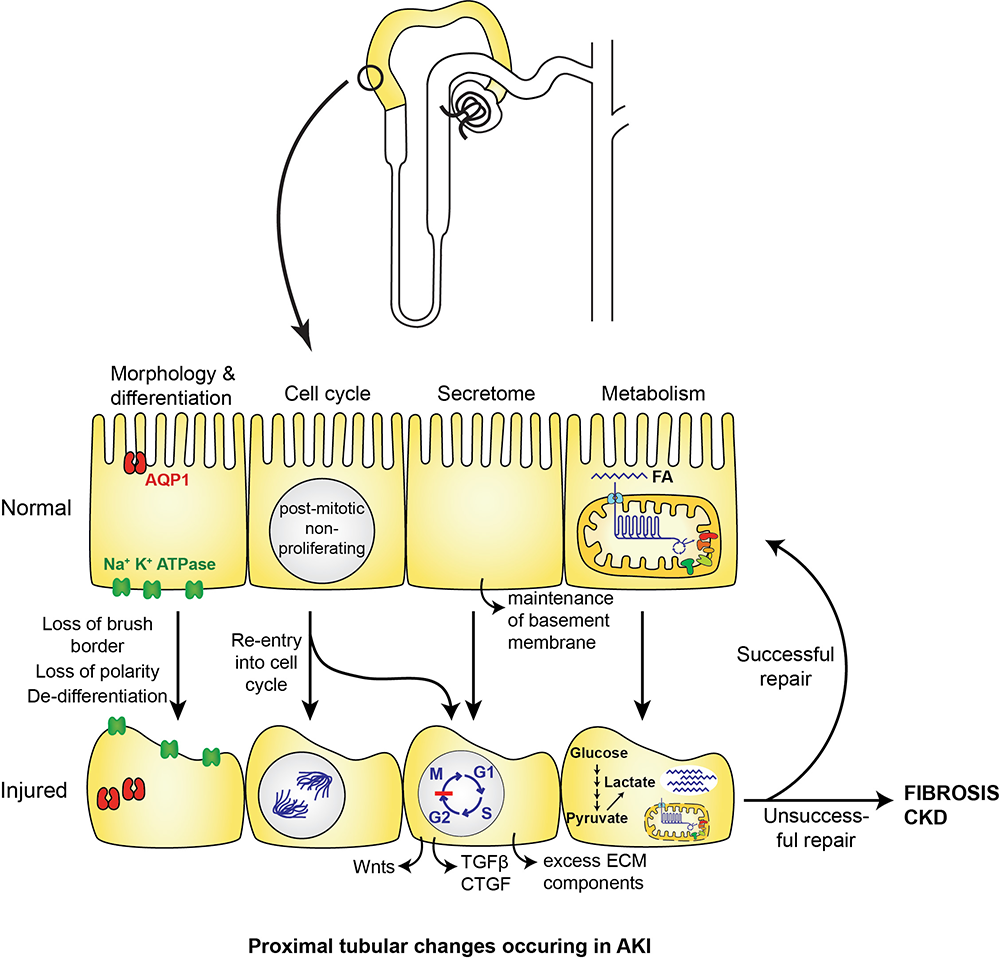
Project 1: Role of KLF6 and KLF15 in control of PT cellular metabolism
Our previous data has shown that KLF6 is likely to transcriptionally repress expression of genes involved in branched chain amino acid (BCAA; Val, Leu, Ile) catabolism. and that KLF15 likely cooperates with PPARα to control fatty acid oxidation (FAO). We are studying the mechanisms by which KLF6 and KLF15 may co-operate to regulate BCAA catabolism and FAO, either directly and/or through mTORC1 signaling.
Project 2: Role of BCAA catabolism in severity of and recovery after AKI
BCAA are catabolized in the kidney to generate TCA cycle intermediates, but the significance of these metabolic pathways for kidney function have not previously been explored. We have shown that expression of genes encoding BCAA catabolic enzymes is reduced in various forms of AKI and CKD. We now aim to understand the significance of BCAA catabolism in AKI by undertaking studies in vitro and in vivo to determine whether loss of BCAA catabolism results in increased susceptibility to AKI and/or CKD; and whether activation of BCAA catabolism can protect against AKI and/or CKD.
Project 3: Role of RNA polymerase II in kidney responses to DNA damage in AKI and CKD
We recently showed that the gene encoding the largest subunit of RNA polymerase II, POLR2A, was upregulated in injured PT cells after DNA damage. Over time, the corresponding protein, RPB1, became increasingly associated with de-differentiated PT cells, which likely propagate fibrosis. When RNA polymerase II encounters DNA damage during transcription, it may become stalled. We are studying mechanisms by which POLR2A/RPB1 is upregulated in AKI, and the effects of stalled RNA polymerase II in driving PT injury and transition to fibrosis after AKI.
KEY PUBLICATIONS
Piret SE*, DiMartino S, Hanubal M, Davis M, Wang J, Bahadur T, Rath A, Gujarati NA, Frimpong BO, Bronstein R, Guo Y, Mallipattu SK*. Krüppel-like factor 6 induces RNA polymerase II subunit RPB1 to promote kidney injury. J Am Soc Nephrol (2025) 36: 1914-1927. *joint corresponding authors.
Accompanying editorial: J Am Soc Nephrol (2025) 36: 1891-1893
DiMartino S, Revelo MP, Mallipattu SK, Piret SE. Activation of branched chain amino acid catabolism protects against nephrotoxic acute kidney injury. Am J Physiol Renal Physiol (2025) 328: F152-F163.
Accompanying editorial: Am J Physiol Renal Physiol (2025) 328: F596-F598
Piret SE, Guo Y, Attallah AA, Horne SJ, Zollman A, Owusu D, Henein J, Sidorenko VS, Revelo MP, Hato T, Ma’ayan A, He JC, Mallipattu SK. Krüppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. PNAS (2021) 118: e2024414118.
Piret SE*, Attallah AA*, Gu X, Guo Y, Gujarati NA, Henein J, Zollman A, Hato T, Ma’ayan A, Revelo MP, Dickman KG, Chen CH, Shun CT, Rosenquist TA, He JC, Mallipattu SK. Loss of proximal tubular Krüppel-like factor 15 (KLF15) exacerbates kidney injury through loss of fatty acid oxidation. Kidney International (2021) 100: 1250-67. *joint first authors.
Accompanying commentary: Kidney International (2021) 100: 1165-1167
Piret SE, Olinger E, Reed AAC, Nesbit MA, Hough TA, Bentley L, Devuyst O, Cox RD, Thakker RV. Mouse model for inherited renal fibrosis associated with endoplasmic reticulum stress. Disease Models and Mechanisms (2017) 10: 773-86.
Full list of publications: NIH Publications - Piret Bibliography
RECENT FUNDING
NIH-NIDDK 1R01DK133238 (2023-2027): “Role of branched-chain catabolism in the proximal tubule” NIH RePORTER; SBU News
Stony Brook Department of Medicine Pilot Project Grant (2023-2024): “Development of high-throughput assays for screening of novel small molecule therapeutics”
American Heart Association Career Development Award (2022-2025): "Role of branched-chain amino acid catabolism in the proximal tubule"
KidneyCure (American Society of Nephrology) Joseph V. Bonventre Research Scholar Grant (2020-2022)
UAB-UCSD O’Brien Center for Acute Kidney Injury Research Pilot & Feasibility Grant (2018-2020)