Tuberculosis (TB) remains a leading cause of global mortality1, 2, yet the fundamental immune mechanisms underlying innate resistance or susceptibility to initial M. tuberculosis (Mtb) infection remain poorly understood3, 4, 5. In order to achieve global TB eradication, fundamental questions regarding host immunity must be answered, namely (1) why do a subset of those highly exposed to Mtb remain uninfected? and (2) How do those who develop latent, asymptomatic infection control latency?
Increasing evidence suggest that rapid immune responses at mucosal surfaces by innate-like lymphocytes that express highly conserved T cell receptors (TCR) recognizing non-peptide microbial ligands may contribute to early Mtb control and clearance prior to priming of adaptive immunity6, 7. The central scientific focus of my laboratory is to identify and target mechanisms of innate lymphocyte immunity against Mtb infection using systems immunology approaches in human tuberculosis cohorts and mouse models of infection.
My research program has three objectives:
1) Identify innate lymphocyte immune mechanisms contributing to resistance to initial Mtb infection.
2) Distinguish innate lymphocyte immune responses induced by tuberculosis latency and active disease.
3) Target innate lymphocytes in vivo as immunotherapy and preventive vaccination.
These proposed studies to investigate and harness innate lymphocyte immunity as host-directed immunotherapy against tuberculosis are attractive as existing duration and toxicities of anti-TB pharmacotherapy regimens result in poor adherence, relapse, and drug-resistance1. Additionally, immune correlates of protection against Mtb infection remain elusive and previous vaccine studies have largely focused on conventional, peptide-specific T cells with limited success8. This proposed work will make significant contributions to identify novel innate lymphocyte targets for therapeutics against Mtb infection.
Building on systems I established as a postdoctoral fellow and ongoing work as a junior faculty, answers to these fundamental questions of host immunity are within reach. In collaboration with the Aubé lab (UNC) and the NIH tetramer Core facility (Emory, GA), we developed new tools to study the abundant innate-like T cell, mucosal-associated invariant T (MAIT) cells, that respond to Vitamin B intermediates derived from microbes when presented by the oligomorphic MHC Ib-related protein, MR1. I characterized the MR1 precursor ligand 5-amino-6- D-ribitylaminouracil (5-A-RU) newly stabilized as a hydrochloride salt9. We harnessed this chemistry to produce abundant stable compound, which could then be reacted with methylglyoxal (MeG) in vitro to form the potent MR1 ligand 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU). Our work facilitated the development of MR1-5-OP-RU tetramers to identify MAIT cells in humans and mice by flow cytometry now available to any interested investigator through the NIH Tetramer Core. Further, I used this purified pre-cursor ligand to develop novel in vitro antigen-specific assays to interrogate MAIT cell function in clinical samples and murine models. We are also actively synthesizing and screening novel 5-A-RU and 5-OP-RU analogs to enhance MAIT cell targeting and to better characterize MR1 ligand diversity.
In order to understand the early immune responses induced by initial Mtb exposure, we recruited a cohort of recently exposed healthy household contacts of active TB patients as well as unexposed community controls in collaboration with Daniel Fitzgerald (WCM) and the GHESKIO centers, Port-au-Prince, Haiti (NIH grant # 1 U19 AI111143) (Fig. 1). IGRA (Mtb-specific interferon g release assay) was used as a clinical biomarker to distinguish exposed contacts who remain uninfected (IGRA-) from contacts with latent TB infection (IGRA+; LTBI). Flow cytometric analysis of peripheral blood mononuclear cells (PBMCs) of contacts compared to controls matched by IGRA status revealed that MAIT and gd T cells respond to initial Mtb exposure and infection, demonstrating distinct phenotypes associated with CD4 or CD8 co-expression in each subset4. Further, we found that MAIT cells demonstrated preserved granzyme B (GZMB) and suppressed IFNg responses in contacts who resisted initial infection (IGRA-). Based on these initial findings, I hypothesized that MAIT cells could be protective against tuberculosis. I subsequently used purified MR1 ligand+TLR2/6 agonist (Pam2Cys; P2C) intranasal costimulation in vivo to test MAIT cell priming in lung as a protective strategy against murine Mtb aerosol challenge. In a recent publication, I reported that MR1 ligand vaccination prior to aerosol challenge induced robust activation and enrichment of MAIT cells in murine lung, but was not sufficient to protect against infection,10 suggesting that additional co-stimulatory signals may be required to enhance bactericidal activity.
Based on these initial findings, I sought to further characterize MAIT cell functional heterogeneity by integrating single cell RNA sequencing with flow cytometric validation in healthy donors. Using this systems immunology approach, we were able to construct the largest MAIT cell transcriptional atlas to date spanning homeostatic and activated states11. I propose to expand this work to identify MAIT, gd T and Natural Killer cell clones induced during initial Mtb exposure and infection in our contact cohort as well as genetically distinct populations with high TB endemicity. My overarching goal is to apply these interdisciplinary approaches to identify candidate innate lymphocyte targets for immunotherapy against Mtb 7.
PAST AND CURRENT RESEARCH
Project I: MAIT cell immunity during human Mtb exposure and infection
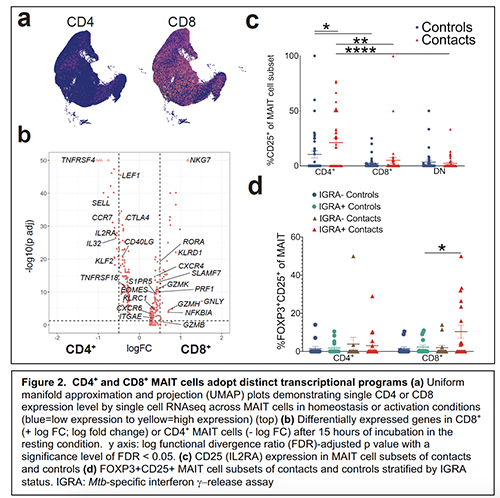 My laboratory seeks to characterize the transcriptional and functional heterogeneity within innate lymphocyte subsets after initial Mtb exposure. To this end, I performed single-cell RNA sequencing of over 76,000 MAIT cells sorted from PBMCs of healthy donors that were incubated in resting and activated conditions in collaboration with the Leslie lab (MSKCC) and constructed a transcriptional atlas of MAIT cell states11. This study provides a comprehensive description of distinct MAIT cell functions adopted during acute and chronic antigen-specific stimulation as contrasted with homeostatic conditions or non-specific T cell activation with aCD3/CD28. We show that CD4+ and CD8+ MAIT cells execute distinct transcriptional programs (Fig. 2a,b) with differential expression of TNFRSF4 and TNFRSF18 in CD4+ MAIT cells suggesting that this subset requires unique costimulatory conditions. MAIT cells also express additional markers of interest in disease states, such as activation/exhaustion markers PD1 and LAG3, as well as the regulatory molecule FOXP3, raising several new hypotheses and therapeutic implications. Moreover, flow cytometric validation in our household contact cohort including CD25 (IL2RA) and FOXP3 (Fig. 2c,d) validate the in vivo relevance of these CD4+ and CD8+ MAIT cell subsets during early Mtb exposure—uncovered for the first time by single cell transcriptomics. Our analysis also reveals an unanticipated functional heterogeneity in MAIT cells that belies their present conception as simple effectors of elimination of bacterially infected cells. For example, while most MAIT cells are driven towards a cytotoxic, IFNg/TNF/GZB+ state after acute activation, we observed surprisingly diverse states adopted during chronic activation. Notably, FOXP3 expression was upregulated during acute and chronic antigen-specific activation and was detected at higher levels in recently infected household contacts of TB cases (Fig. 2d). These findings motivate further characterization of these MAIT cell subsets during initial Mtb exposure and infection.
My laboratory seeks to characterize the transcriptional and functional heterogeneity within innate lymphocyte subsets after initial Mtb exposure. To this end, I performed single-cell RNA sequencing of over 76,000 MAIT cells sorted from PBMCs of healthy donors that were incubated in resting and activated conditions in collaboration with the Leslie lab (MSKCC) and constructed a transcriptional atlas of MAIT cell states11. This study provides a comprehensive description of distinct MAIT cell functions adopted during acute and chronic antigen-specific stimulation as contrasted with homeostatic conditions or non-specific T cell activation with aCD3/CD28. We show that CD4+ and CD8+ MAIT cells execute distinct transcriptional programs (Fig. 2a,b) with differential expression of TNFRSF4 and TNFRSF18 in CD4+ MAIT cells suggesting that this subset requires unique costimulatory conditions. MAIT cells also express additional markers of interest in disease states, such as activation/exhaustion markers PD1 and LAG3, as well as the regulatory molecule FOXP3, raising several new hypotheses and therapeutic implications. Moreover, flow cytometric validation in our household contact cohort including CD25 (IL2RA) and FOXP3 (Fig. 2c,d) validate the in vivo relevance of these CD4+ and CD8+ MAIT cell subsets during early Mtb exposure—uncovered for the first time by single cell transcriptomics. Our analysis also reveals an unanticipated functional heterogeneity in MAIT cells that belies their present conception as simple effectors of elimination of bacterially infected cells. For example, while most MAIT cells are driven towards a cytotoxic, IFNg/TNF/GZB+ state after acute activation, we observed surprisingly diverse states adopted during chronic activation. Notably, FOXP3 expression was upregulated during acute and chronic antigen-specific activation and was detected at higher levels in recently infected household contacts of TB cases (Fig. 2d). These findings motivate further characterization of these MAIT cell subsets during initial Mtb exposure and infection.
My independent lab will perform single cell RNA sequencing of MAIT cells in recently exposed TB household contacts and active TB cases compared to IGRA-matched community controls. We will then map MAIT cell clusters identified during Mtb exposure and infection onto our existing reference MAIT cell atlas generated by single cell profiling of healthy donors11. This will not only validate the generalizability of our current transcriptional atlas, but also determine how MAIT cell effector programs diverge from homeostatic states during early Mtb exposure. I will also perform parallel TCR sequencing (TCRseq) of MR1-tetramer+ cells in these cohorts, which is particularly poignant as 1-10% of MR1 tetramer-reactive cells do not express TRAV1-2 (data not shown) and may represent distinct MR1-reactive populations with different ligand avidity and costimulatory requirements. Moreover, previous TCRseq studies of human MAIT cells either omitted CD4 expressing cells in the experimental design or relied on monoclonal antibodies against a TCR Va7.2 that also bind to TRAV1-2+ non-MAIT conventional T cells (up to 20% in some donors; data not shown), rather than MR1 tetramers12, 13. This single cell genomics approach is complemented by ongoing in vitro functional studies to understand the in vivo conditions selecting for MAIT cell subset specialization (eg, TNFRSF4/18 ligands as costimulatory molecules for CD4+ MAIT cell expansion; FOXP3 expressing MAIT cells as inhibitors of T cell proliferation; checkpoint molecules such as LAG3 and inhibition of MAIT cell clonal expansion). This project will identify MAIT cell clones induced by initial Mtb exposure, early latent infection, and active TB disease and determine MAIT cell-directed targets against tuberculosis.
Project II: NK cell immunity during human Mtb exposure and infection
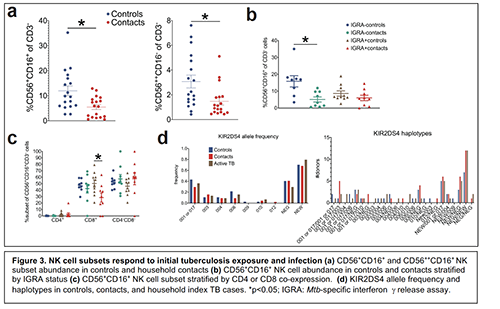 My initial work on MAIT cell immunity and TB has expanded to interrogate the canonical innate lymphoid subsets that are also hypothesized to respond during initial tuberculosis infection14. My current focus is on the most abundant of these innate lymphocytes in peripheral blood—Natural Killer (NK) cells—that recognize diverse ligands through pattern recognition receptors (PRRs) and Killer immunoglobulin receptors (KIRs)15, 16. KIRs demonstrate an impressive degree of allelic diversity allowing them to bind MHC Class I (HLA I) ligands and to detect malignantly transformed or virally infected cells16. Emerging evidence demonstrates that NK cells also recognize bacterial epitopes17. In my preliminary flow cytometric characterization of NK cells in our TB contact cohorts, I observed decreased relative abundance of CD56++CD16- and CD56+CD16+ NK cells in contacts vs unexposed community controls (Fig 3a). When stratifying CD56+CD16+ NK cells by IGRA status, this depletion was most notable in uninfected contacts (IGRA-) (Fig 3a). After further stratifying by CD4 or CD8 co-expression, the relative expression of CD8+ CD56+CD16+ NK cells was decreased in latently infected (IGRA+) contacts (Fig. 3c). These results suggest that distinct NK cell subsets respond during initial Mtb exposure and early LTBI and that these responses are distinguished by NK cell CD8 expression. Ongoing work will validate these findings in a larger sampling from the same Haitian cohorts and will include active TB patients to also identify NK cell correlates of disease progression. Plasma has also been collected for cytomegalovirus (CMV) antibody screening, as CMV infection status drives avidity selection of NK cell memory subsets18, which we hypothesize enhance NK cell immune responses against Mtb. Additionally, this project will sequence KIR and HLA receptors from whole blood DNA of these same donors in collaboration with the Hsu lab (MSKCC) and DKMS laboratories (Dresden, Germany)19, 20. We will use multiplex, parallel KIR-HLA gene sequencing to test the hypothesis that specific haplotypes are enriched in donors who are exposed, but remain uninfected (IGRA-), that may represent an NK cell-mediated resistance mechanism against initial infection. Similarly, haplotypes enriched in latently infected donors or active TB cases may provide clues as to KIR-mediated mechanisms of susceptibility to infection21, 22. Our preliminary analyses in 150 donors have identified novel single nucleotide polymorphisms (SNPs) in previously described KIR alleles such as KIR2DS4 (Fig. 3d), highlighting the importance of expanding our current genetic libraries to include underrepresented minority populations such as in Haiti. Moreover, we observed that some of these SNPs are enriched in households with Mtb transmission. This project will not only impact the field of KIR immunogenetics, but also identify candidate KIR-HLA haplotypes that can be screened for bactericidal activity in vitro. I propose to do this using PBMCs from our existing biobanks with known HLA-KIR haplotypes (Hsu lab) in Mtb-infected autologous monocyte cell culture models I have developed. This project will identify the immunologic and genetic correlates of NK cell-mediated immunity during initial Mtb exposure and infections.
My initial work on MAIT cell immunity and TB has expanded to interrogate the canonical innate lymphoid subsets that are also hypothesized to respond during initial tuberculosis infection14. My current focus is on the most abundant of these innate lymphocytes in peripheral blood—Natural Killer (NK) cells—that recognize diverse ligands through pattern recognition receptors (PRRs) and Killer immunoglobulin receptors (KIRs)15, 16. KIRs demonstrate an impressive degree of allelic diversity allowing them to bind MHC Class I (HLA I) ligands and to detect malignantly transformed or virally infected cells16. Emerging evidence demonstrates that NK cells also recognize bacterial epitopes17. In my preliminary flow cytometric characterization of NK cells in our TB contact cohorts, I observed decreased relative abundance of CD56++CD16- and CD56+CD16+ NK cells in contacts vs unexposed community controls (Fig 3a). When stratifying CD56+CD16+ NK cells by IGRA status, this depletion was most notable in uninfected contacts (IGRA-) (Fig 3a). After further stratifying by CD4 or CD8 co-expression, the relative expression of CD8+ CD56+CD16+ NK cells was decreased in latently infected (IGRA+) contacts (Fig. 3c). These results suggest that distinct NK cell subsets respond during initial Mtb exposure and early LTBI and that these responses are distinguished by NK cell CD8 expression. Ongoing work will validate these findings in a larger sampling from the same Haitian cohorts and will include active TB patients to also identify NK cell correlates of disease progression. Plasma has also been collected for cytomegalovirus (CMV) antibody screening, as CMV infection status drives avidity selection of NK cell memory subsets18, which we hypothesize enhance NK cell immune responses against Mtb. Additionally, this project will sequence KIR and HLA receptors from whole blood DNA of these same donors in collaboration with the Hsu lab (MSKCC) and DKMS laboratories (Dresden, Germany)19, 20. We will use multiplex, parallel KIR-HLA gene sequencing to test the hypothesis that specific haplotypes are enriched in donors who are exposed, but remain uninfected (IGRA-), that may represent an NK cell-mediated resistance mechanism against initial infection. Similarly, haplotypes enriched in latently infected donors or active TB cases may provide clues as to KIR-mediated mechanisms of susceptibility to infection21, 22. Our preliminary analyses in 150 donors have identified novel single nucleotide polymorphisms (SNPs) in previously described KIR alleles such as KIR2DS4 (Fig. 3d), highlighting the importance of expanding our current genetic libraries to include underrepresented minority populations such as in Haiti. Moreover, we observed that some of these SNPs are enriched in households with Mtb transmission. This project will not only impact the field of KIR immunogenetics, but also identify candidate KIR-HLA haplotypes that can be screened for bactericidal activity in vitro. I propose to do this using PBMCs from our existing biobanks with known HLA-KIR haplotypes (Hsu lab) in Mtb-infected autologous monocyte cell culture models I have developed. This project will identify the immunologic and genetic correlates of NK cell-mediated immunity during initial Mtb exposure and infections.
Project III: Targeting innate lymphocyte immunity in vivo
In this project, I propose to target MAIT and NK cells in vivo as prevention or treatment immunotherapy against Mtb. These approaches will use antigen-specific or cytokine-based mucosal vaccination strategies in an effort to enhance MAIT and NK cell activation and proliferation in murine lung prior to or after Mtb aerosol challenge. While we and other groups have recently demonstrated that MAIT cell enrichment in lung is not sufficient to protect against Mtb infection23, 24, 25, emerging data demonstrate that Mtb-specific MAIT cell responses may be more dependent on IL12/IL18 than MR126 and that MAIT cell enrichment in murine lung is IL23-dependent27. It is also possible that MAIT cells are inhibited by a combination of MR1 ligands unique to Mtb28. Together, these studies suggest that MAIT cells may require additional co-stimulatory signals not conferred by TLR ligand/5-OP-RU alone to enhance bactericidal activity. One recent study demonstrated ~1 log-fold decrease in Mtb lung bacterial load after serial 5-OP-RU priming during advanced murine infection25. This data raises the possibility that MAIT cell-directed therapy could be used as treatment of chronic disease. My preliminary work demonstrates that intranasal boosting with P2C/5OPRU during chronic infection further enriches for MAIT cells in murine lung (Fig. 4a) and I plan to test this treatment strategy in murine models of post-exposure prophylaxis after acute infection and treatment of chronic infection, either alone or as adjuvants to existing anti-TB pharmacotherapy. In addition, intranasal inoculation of IL23/5-OP-RU also enhances MAIT cell numbers in lung in the absence of TLR co-stimulation (Fig. 4b) and may provide additional co-stimulatory signals that enhance MAIT cell-directed immunity against Mtb. Moreover, I have ongoing collaborations with the Aubé lab (UNC) to synthesize and screen novel 5-A-RU/5-OP-RU analogs that activate human and murine MAIT cells in vitro and in vivo, some of which have activity without the addition of methylglyoxal, and demonstrate strikingly varied effects on MAIT cell proliferation (Fig. 4c,d).
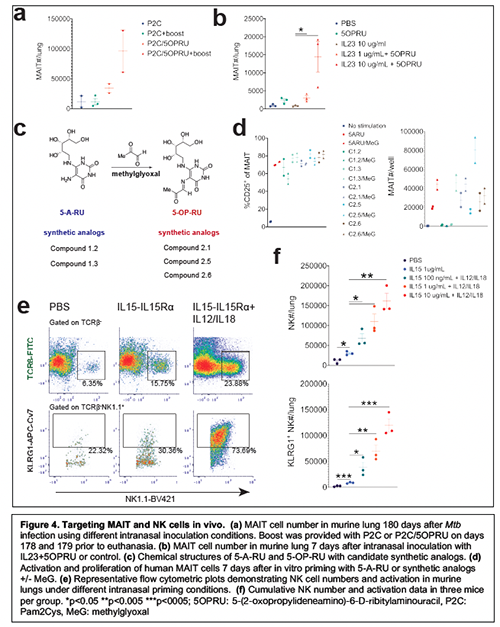 Identifying compounds that directly bind to MR1 and modulate MAIT cell activation without the addition of methylglyoxal will facilitate more precise in vitro/in vivo dose titration as well as enhance accuracy of our iterative structure activity analyses. These ongoing studies will identify candidate synthetic MR1 ligands for in vivo delivery and can further our understanding of MR1-TCR interactions to identify selective ligands for CD4+ or CD8+ MAIT cells. These studies also seek to identify hypothesized Mtb-specific MR1 ligands28 as well as MR1 self ligands29. To target NK cells, I have developed an in vivo model of mucosal priming using IL15-IL15Ra fusion constructs with targeted mutations in the IL15 moiety or truncations of the IL15 receptor alpha chain (IL15Ra) in collaboration with the Cheung lab (MSKCC). IL15 induces NK cell differentiation and expansion, can be presented in trans to IL2R/IL15Rb by IL15Ra, and is currently in use to expand NK cells in cancer immunotherapy trials30, 31. Importantly, IL15-deficient mice have impaired Mtb control resulting in increased bacterial loads and decreased survival during chronic infection32. While NK cells respond to murine aerosol Mtb infection, in vivo depletion of NK cells early after aerosol challenge did not affect bacterial load33, suggesting that these cells play a minimal role in protective immunity in mice. However, few efforts have been made to directly target NK cells as immunotherapy against tuberculosis34. I have established a model of intranasal vaccination with IL15-IL15Ra (wild type IL15-truncated IL15Ra; Sushi domain)35 +/- IL12/IL18 (Fig. 4e,f), which induces robust enrichment and activation of NK cells in murine lung. I propose to test IL15 fusion constructs (Cheung lab) as protective or therapeutic mucosal vaccination strategies against Mtb in vivo using our well-established murine aerosol infection models10.
Identifying compounds that directly bind to MR1 and modulate MAIT cell activation without the addition of methylglyoxal will facilitate more precise in vitro/in vivo dose titration as well as enhance accuracy of our iterative structure activity analyses. These ongoing studies will identify candidate synthetic MR1 ligands for in vivo delivery and can further our understanding of MR1-TCR interactions to identify selective ligands for CD4+ or CD8+ MAIT cells. These studies also seek to identify hypothesized Mtb-specific MR1 ligands28 as well as MR1 self ligands29. To target NK cells, I have developed an in vivo model of mucosal priming using IL15-IL15Ra fusion constructs with targeted mutations in the IL15 moiety or truncations of the IL15 receptor alpha chain (IL15Ra) in collaboration with the Cheung lab (MSKCC). IL15 induces NK cell differentiation and expansion, can be presented in trans to IL2R/IL15Rb by IL15Ra, and is currently in use to expand NK cells in cancer immunotherapy trials30, 31. Importantly, IL15-deficient mice have impaired Mtb control resulting in increased bacterial loads and decreased survival during chronic infection32. While NK cells respond to murine aerosol Mtb infection, in vivo depletion of NK cells early after aerosol challenge did not affect bacterial load33, suggesting that these cells play a minimal role in protective immunity in mice. However, few efforts have been made to directly target NK cells as immunotherapy against tuberculosis34. I have established a model of intranasal vaccination with IL15-IL15Ra (wild type IL15-truncated IL15Ra; Sushi domain)35 +/- IL12/IL18 (Fig. 4e,f), which induces robust enrichment and activation of NK cells in murine lung. I propose to test IL15 fusion constructs (Cheung lab) as protective or therapeutic mucosal vaccination strategies against Mtb in vivo using our well-established murine aerosol infection models10.
FUTURE DIRECTIONS
My long-term goal is to harness the conditions selective for Mtb-specific MAIT and NK cell clonal expansion to develop small molecule activators/inhibitors that enhance bactericidal activity and modulate pathologic inflammation (Fig. 5a,b). Future aims include development of novel co-stimulatory cocktails for MAIT and NK-directed therapy to prevent Mtb infection, sterilize latent infection, or treat chronic disease.
To this end, my laboratory seeks to expand current Global Health Center collaborations to study innate lymphocyte responses to early Mtb exposure and infection in genetically distinct populations, including acquiring samples from TB research units with ongoing recruitment of household contact cohorts and longitudinal follow-up of healthy donors in high Mtb transmission settings (Fig. 5c). These additional collaborations will be critical to assessing the generalizability of our findings in Haiti as well as to assess therapeutic interventions in diverse populations.
Moreover, my systems immunology approach is highly applicable to other infectious and non-infectious immune-mediated disease states such as cancer and autoimmunity where MR1-reactive T and NK cell subsets are rapidly emerging as attractive cellular targets for immunotherapy13, 29, 36, 37 (Fig. 5d). Once my laboratory is established, my eventual goal will be to widen my focus and initiate new clinical translational collaborations to interrogate MR1 reactive T and NK cells in major TB co-morbidities such as HIV and malaria, as well as non-communicable, immune-mediated diseases.
CONCLUDING REMARKS
My postdoctoral training and ongoing work as junior faculty resulted in: 1) the development of novel antigen-specific MAIT cell assays to interrogate human and murine MAIT cells during Mtb infection (2) identification of CD4+ and CD8+ MAIT, gd T and NK cell correlates of initial Mtb exposure and infection in household contacts of active TB patients (3) development of MR1 ligand and IL15 fusion construct vaccination models to assess MAIT and NK cell-directed therapy against tuberculosis in vivo and (4) the construction of a single cell transcriptional atlas of MAIT cell homeostasis and activation to serve as a benchmark for interrogation of MAIT clusters in disease states. This body of work establishes the foundation for my proposed studies focusing on early innate lymphocyte responses against Mtb. I expect to initiate projects I and II shortly after starting my lab while building towards animal models described in project III. My proposed research will identify innate lymphocyte phenotypes contributing to resistance/susceptibility to Mtb infection and reveal novel host-directed targets for anti-TB immunotherapy and mucosal vaccine development.
REFERENCES
1. WHO. WHO Global Tuberculosis Report 2020. (2020).
2. Vorkas, C. et al. Tuberculosis drug resistance and outcomes among tuberculosis inpatients in Lilongwe, Malawi. Malawi Med J 24, 21-24 (2012)
3. Cassidy, J.P. & Martineau, A.R. Innate resistance to tuberculosis in man, cattle and laboratory animal models: nipping disease in the bud? J Comp Pathol 151, 291-308 (2014).
4. Vorkas, C.K. et al. Mucosal-associated invariant and gammadelta T cell subsets respond to initial Mycobacterium tuberculosis infection. JCI Insight 3 (2018).
5. Coulter, F. et al. IL-17 Production from T Helper 17, Mucosal-Associated Invariant T, and gammadelta Cells in Tuberculosis Infection and Disease. Front Immunol 8, 1252 (2017).
6. Nemes, E., Khader, S.A., Swanson, R.V. & Hanekom, W.A. Targeting Unconventional Host Components for Vaccination-Induced Protection Against TB. Front Immunol 11, 1452 (2020).
7. Vorkas, C.K. & Glickman, M.S. MAIT and gd T cells. Advances in host-directed therapies against tuberculosis (2020).
8. Satti, I. & McShane, H. Current approaches toward identifying a correlate of immune protection from tuberculosis. Expert Rev Vaccines 18, 43-59 (2019).
9. Li, K. et al. Synthesis, stabilization, and characterization of the MR1 ligand precursor 5-amino-6-Dribitylaminouracil (5-A-RU). PLoS One 13, e0191837 (2018).
10. Vorkas, C.K. et al. Efficient 5-OP-RU-induced enrichment of Mucosal-associated invariant T cells in the murine lung does not enhance control of aerosol Mycobacterium tuberculosis infection. bioRxiv (2020).
11. Vorkas, C.K. et al. Single cell transcriptional profiling reveals helper, effector, and regulatory MAIT cell populations enriched during homeostasis and activation. bioRxiv (2020).
12. Lepore, M. et al. Parallel T-cell cloning and deep sequencing of human MAIT cells reveal stable oligoclonal TCRbeta repertoire. Nat Commun 5, 3866 (2014).
13. Carnero Contentti, E., Farez, M.F. & Correale, J. Mucosal-Associated Invariant T Cell Features and TCR Repertoire Characteristics During the Course of Multiple Sclerosis. Front Immunol 10, 2690 (2019).
14. Verrall, A.J., Netea, M.G., Alisjahbana, B., Hill, P.C. & van Crevel, R. Early clearance of Mycobacterium tuberculosis: a new frontier in prevention. Immunology 141, 506-513 (2014).
15. Hsu, K.C. et al. Killer Ig-like receptor haplotype analysis by gene content: evidence for genomic diversity with a minimum of six basic framework haplotypes, each with multiple subsets. J Immunol 169, 5118-5129 (2002).
16. Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5, 201-214 (2005).
17. Sim, M.J.W. et al. Human NK cell receptor KIR2DS4 detects a conserved bacterial epitope presented by HLA-C. Proc Natl Acad Sci U S A 116, 12964-12973 (2019).
18. Adams, N.M. et al. Cytomegalovirus Infection Drives Avidity Selection of Natural Killer Cells. Immunity 50, 1381-1390 e1385 (2019).
19. Wagner, I. et al. Allele-Level KIR Genotyping of More Than a Million Samples: Workflow, Algorithm, and Observations. Front Immunol 9, 2843 (2018).
20. Le Luduec, J.B., Kudva, A., Boudreau, J.E. & Hsu, K.C. Novel multiplex PCR-SSP method for centromeric KIR allele discrimination. Sci Rep 8, 14853 (2018).
21. Pydi, S.S. et al. Killer cell immunoglobulin like receptor gene association with tuberculosis. Hum Immunol 74, 85-92 (2013).
22. Braun, K. et al. Killer immunoglobulin-like receptor (KIR) centromeric-AA haplotype is associated with ethnicity and tuberculosis disease in a Canadian First Nations cohort. PLoS One 8, e67842 (2013).
23. Vorkas, C.K. et al. Efficient 5-OP-RU-induced enrichment of Mucosal-associated invariant T cells in the murine lung does not enhance control of aerosol Mycobacterium tuberculosis infection. Infect Immun (2020).
24. Yu, H. et al. Artificially induced MAIT cells inhibit M. bovis BCG but not M. tuberculosis during in vivo pulmonary infection. Sci Rep 10, 13579 (2020).
25. Sakai, S. et al. MAIT cell-directed therapy of Mycobacterium tuberculosis infection. Mucosal Immunol (2020).
26. Suliman, S. et al. MR1-Independent Activation of Human Mucosal-Associated Invariant T Cells by Mycobacteria. J Immunol 203, 2917-2927 (2019).
27. Wang, H. et al. IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci Immunol 4 (2019).
28. Harriff, M.J. et al. MR1 displays the microbial metabolome driving selective MR1-restricted T cell receptor usage. Sci Immunol 3 (2018).
29. Crowther, M.D. et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol 21, 178-185 (2020).
30. Lauwerys, B.R., Garot, N., Renauld, J.C. & Houssiau, F.A. Cytokine production and killer activity of NK/T-NK cells derived with IL-2, IL-15, or the combination of IL-12 and IL-18. J Immunol 165, 1847-1853 (2000).
31. Conlon, K.C. et al. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin Cancer Res 25, 4945-4954 (2019).
32. Rausch, A. et al. Interleukin-15 mediates protection against experimental tuberculosis: a role for NKG2D-dependent effector mechanisms of CD8+ T cells. Eur J Immunol 36, 1156-1167 (2006).
33. Junqueira-Kipnis, A.P. et al. NK cells respond to pulmonary infection with Mycobacterium tuberculosis, but play a minimal role in protection. J Immunol 171, 6039-6045 (2003).
34. Tang, C. et al. Efficacy of recombinant bacille Calmette-Guerin vaccine secreting interleukin-15/antigen 85B fusion protein in providing protection against Mycobacterium tuberculosis. J Infect Dis 197, 1263-1274 (2008).
35. Mortier, E. et al. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem 281, 1612-1619 (2006).
36. Gherardin, N.A. et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep 8, 4159 (2018).
37. Yan, J. et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov 10, 124-141 (2020).